An overview of GW and its applications to core level spectroscopy
Dr. Ramón L. Panadés Barrueta
panadestein.github.io
Why is it called GW anyway?
GW in a nutshell
- Perturbative methods for solving the many-body problem.
The main object in the method is the one-particle Green's function:
\begin{equation*} \LARGE{G(\mathbf{r}, \mathbf{r'}; \omega) = \sum_n \frac{f_n(\mathbf{r})f_n^{*}(\mathbf{r'})} {\hbar\omega - \epsilon_n +i\eta\hbar\text{sgn}(\epsilon_n-\mu)}} \end{equation*}- Excellent for charge excitations (IPs, EAs, fundamental gaps, CE)
- Can be used for finite size or extended systems, up to hundreds of atoms.
Derivation
GW is an approximation to an exact set of integro-differential equations (Hedin's equations). The latter can be derived using a diagrammatic approach, or alternatively the Schwinger's functional derivative technique.

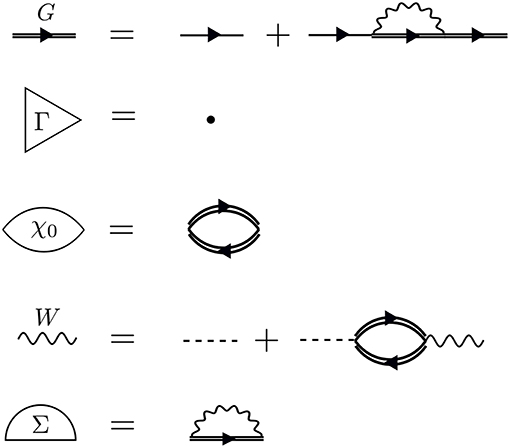
Image from GW compendium
From Hedin's equations to GW


Images from ABINIT website
Practical GW calculations
Quasiparticle equation
Starting from Dyson's equation in differential form:
Taking the Fourier transform to the energy domain and using the eigenvalue expansion of the Green's function:
Typical G0W0 algorithm

GW typically scales as \(\color{red}{\mathbf{O(N^4)}}\), we are working on reducing the scaling to \(\color{green}{\mathbf{O(N^3)}}\)
The contour deformation approach for the self-energy
A clever application of Jordan's Lemma and Cauchy's residue theorem. Integral self-energy expression is a consequence of the Convolution theorem.


Example application to core level spectroscopy
Software packages implementing GW methods



|



|
See Table 1 in the GW compendium for a complete list
Computation of the spectral function of the H2O 1s orbital
- Showcase FHI-aims workflow with a small application
The contour deformation technique has a larger scaling for core levels \(\color{red}{O(N^5)}\) due to the number of residues:
\begin{equation*} \color{red}{N_{res}N_{occ}N_{virt}N^2_{aux}} \qquad \color{blue}{N_{\omega}N_{occ}N_{virt}N^2_{aux}} \end{equation*}- Description of satellite structure is expensive due to spectral function
The geometry.in file
atom 0.00000000 -0.00000000 -0.00614048 O
atom 0.76443318 -0.00000000 0.58917024 H
atom -0.76443318 0.00000000 0.58917024 H
The control.in file
####################################################
# H2O molecule calculation
# Using def2-QVPZ basis for light and heavy elements
####################################################
#-------------------General options----------------------------
xc pbe # DFT functional
hybrid_xc_coeff 0.45 # Exchange mixing parameter
qpe_calc gw # Single shot GoWo
KS_method serial # Solve the geneigenval problem serial algorithm
override_illconditioning .true. # Override safe options for overlap matrix
RI_method v # Resolution of the identity for GW
prodbas_threshold 1.e-5 # Prevent ill-conditioning of auxiliary basis set
spin none # No spin specified, why?
partition_type rho_r2 # Patition type for integration grids
occupation_type gaussian 0.000001 # Broadening scheme used to find Fermi level
empty_states 25000 # Kohn-Sham states beyond the occupied levels
density_update_method density_matrix # Density matrix updating scheme
sc_accuracy_rho 1E-5 # SCF options
sc_accuracy_eev 1E-5
sc_accuracy_etot 1E-5
sc_iter_limit 400
#--------------------------------------------------------------
#-------------------GW specific options------------------------
anacon_type 1 # Pade analytic continuation
n_anacon_par 16 # Number of parameters in the Pade approximation
frequency_points 200 # Number of imaginary frequency points in \Sigma
contour_def_gw 1 1 # Range of states for which CD is applied
contour_eta 0.002 # Infinitesimal in Lehmann representation
state_lower_limit 1 # Lowest single-particle eigenstate
calc_spectral_func -560 -500 0.001 # Compute spectral function
#--------------------------------------------------------------
#-------------------Relativistic options-----------------------
relativistic none # Non-relativistic calculation
override_relativity .true. # Do not stop the code when questionable input
#--------------------------------------------------------------
#-------------------Density mixing options---------------------
mixer pulay
n_max_pulay 10
charge_mix_param 0.2
#--------------------------------------------------------------
################################################################################
#
# FHI-aims code project
# VB, Fritz-Haber Institut, 2007
#
# Suggested "safe" defaults for H atom (to be pasted into control.in file)
#
################################################################################
species H
include_min_basis false
pure_gauss true
cut_pot 6.0 2.5 1.0
l_hartree 8
basis_dep_cutoff 0.d0
radial_base 100 7.0
radial_multiplier 8
angular_grids auto
angular 1202
angular_acc 1.0e-08
angular_min 110
basis_acc 1.0e-5
# global species definitions
nucleus 1
mass 1.00794
#
#
#
################################################################################
#
# Definition of "minimal" basis
#
################################################################################
# valence basis states
valence 1 s 1.
# ion occupancy
ion_occ 1 s 0.5
################################################################################
#
# Suggested additional basis functions. For production calculations,
# uncomment them one after another (the most important basis functions are
# listed first).
#
# Basis constructed for dimers: 0.5 A, 0.7 A, 1.0 A, 1.5 A, 2.5 A
#
################################################################################
# H cc-pVQZ
gaussian 0 3
82.6400000 0.0020060
12.4100000 0.0153430
2.8240000 0.0755790
gaussian 0 1 0.7977000
gaussian 0 1 0.2581000
gaussian 0 1 0.0898900
gaussian 1 1 2.2920000
gaussian 1 1 0.8380000
gaussian 1 1 0.2920000
gaussian 2 1 2.0620000
gaussian 2 1 0.6620000
gaussian 3 1 1.3970000
################################################################################
#
# FHI-aims code project
# VB, Fritz-Haber Institut, 2007
#
# Suggested "safe" defaults for O atom (to be pasted into control.in file)
#
################################################################################
species O
include_min_basis false
pure_gauss true
cut_pot 6.0 2.5 1.0
l_hartree 8
basis_dep_cutoff 0.d0
radial_base 100 7.0
radial_multiplier 8
angular_grids auto
angular 1202
angular_acc 1.0e-08
angular_min 110
basis_acc 1.0e-5
# global species definitions
nucleus 8
mass 15.9994
#
#
#
################################################################################
#
# Definition of "minimal" basis
#
################################################################################
# valence basis states
valence 2 s 2.
valence 2 p 4.
# ion occupancy
ion_occ 2 s 1.
ion_occ 2 p 3.
################################################################################
#
# Suggested additional basis functions. For production calculations,
# uncomment them one after another (the most important basis functions are
# listed first).
#
# Constructed for dimers: 1.0 A, 1.208 A, 1.5 A, 2.0 A, 3.0 A
#
################################################################################
# O cc-pVQZ
gaussian 0 9
61420.0000000 0.0000900
9199.0000000 0.0006980
2091.0000000 0.0036640
590.9000000 0.0152180
192.3000000 0.0524230
69.3200000 0.1459210
26.9700000 0.3052580
11.1000000 0.3985080
4.6820000 0.2169800
gaussian 0 9
61420.0000000 -0.0000200
9199.0000000 -0.0001590
2091.0000000 -0.0008290
590.9000000 -0.0035080
192.3000000 -0.0121560
69.3200000 -0.0362610
26.9700000 -0.0829920
11.1000000 -0.1520900
4.6820000 -0.1153310
gaussian 0 1 1.4280000
gaussian 0 1 0.5547000
gaussian 0 1 0.2067000
gaussian 1 3
63.4200000 0.0060440
14.6600000 0.0417990
4.4590000 0.1611430
gaussian 1 1 1.5310000
gaussian 1 1 0.5302000
gaussian 1 1 0.1750000
gaussian 2 1 3.7750000
gaussian 2 1 1.3000000
gaussian 2 1 0.4440000
gaussian 3 1 2.6660000
gaussian 3 1 0.8590000
gaussian 4 1 1.8460000
The aims.out file
------------------------------------------------------------
Invoking FHI-aims ...
When using FHI-aims, please cite the following reference:
Volker Blum, Ralf Gehrke, Felix Hanke, Paula Havu,
Ville Havu, Xinguo Ren, Karsten Reuter, and Matthias Scheffler,
'Ab Initio Molecular Simulations with Numeric Atom-Centered Orbitals',
Computer Physics Communications 180, 2175-2196 (2009)
In addition, many other developments in FHI-aims are likely important for
your particular application. A partial list of references is given at the end of
this file. Thank you for giving credit to the authors of these developments.
For any questions about FHI-aims, please visit our slack channel at
https://fhi-aims.slack.com
and our main development and support site at
https://aims-git.rz-berlin.mpg.de .
The latter site, in particular, has a wiki to collect information, as well
as an issue tracker to log discussions, suggest improvements, and report issues
or bugs. https://aims-git.rz-berlin.mpg.de is also the main development site
of the project and all new and updated code versions can be obtained there.
Please send an email to aims-coordinators@fhi-berlin.mpg.de and we will add
you to these sites. They are for you and everyone is welcome there.
------------------------------------------------------------
Date : 20210920, Time : 144849.255
Time zero on CPU 1 : 0.120000000000000E-01 s.
Internal wall clock time zero : 401381329.255 s.
FHI-aims created a unique identifier for this run for later identification
aims_uuid : 1834686C-6A3D-44DB-AA67-5CBBADA1A125
Build configuration of the current instance of FHI-aims
-------------------------------------------------------
FHI-aims version : 210802
Commit number : 8af8a52de
CMake host system : Linux-4.9.0-14-amd64
CMake version : 3.7.2
Fortran compiler : /usr/local/share/intel/parallel_studio_xe_2017/compilers_and_libraries/linux/mpi/intel64/bin/mpiifort (Intel) version 17.0.4.20170411
Fortran compiler flags: -O3 -ip -fp-model precise
C compiler : /usr/local/share/intel/parallel_studio_xe_2017/compilers_and_libraries/linux/bin/intel64/icc (Intel) version 17.0.4.20170411
C compiler flags : -O3 -ip -fp-model precise -std=gnu99
ELPA2 kernel : AVX2
Using MPI
Using ScaLAPACK
Using LibXC
Using i-PI
Using RLSY
Linking against: /usr/local/share/intel/parallel_studio_xe_2017/compilers_and_libraries_2017/linux/mkl/lib/intel64/libmkl_intel_lp64.so
/usr/local/share/intel/parallel_studio_xe_2017/compilers_and_libraries_2017/linux/mkl/lib/intel64/libmkl_sequential.so
/usr/local/share/intel/parallel_studio_xe_2017/compilers_and_libraries_2017/linux/mkl/lib/intel64/libmkl_core.so
/usr/local/share/intel/parallel_studio_xe_2017/compilers_and_libraries_2017/linux/mkl/lib/intel64/libmkl_scalapack_lp64.so
/usr/local/share/intel/parallel_studio_xe_2017/compilers_and_libraries_2017/linux/mkl/lib/intel64/libmkl_blacs_intelmpi_lp64.so
Using 8 parallel tasks.
Task 0 on host cpch06 reporting.
Task 1 on host cpch06 reporting.
Task 2 on host cpch06 reporting.
Task 3 on host cpch06 reporting.
Task 4 on host cpch06 reporting.
Task 5 on host cpch06 reporting.
Task 6 on host cpch06 reporting.
Task 7 on host cpch06 reporting.
Performing system and environment tests:
| Environment variable OMP_NUM_THREADS correctly set to 1.
| Checking for ScaLAPACK...
| Testing pdtran()...
| All pdtran() tests passed.
Obtaining array dimensions for all initial allocations:
-----------------------------------------------------------------------
Parsing control.in (first pass over file, find array dimensions only).
The contents of control.in will be repeated verbatim below
unless switched off by setting 'verbatim_writeout .false.' .
in the first line of control.in .
-----------------------------------------------------------------------
####################################################
# H2O molecule calculation
# Using def2-QVPZ basis for light and heavy elements
####################################################
#-------------------General options----------------------------
xc pbe # DFT functional
qpe_calc gw # Single shot GoWo
KS_method serial # Solve the geneigenval problem serial algorithm
override_illconditioning .true. # Override safe options for overlap matrix
RI_method v # Resolution of the identity for GW
prodbas_threshold 1.e-5 # Prevent ill-conditioning of auxiliary basis set
spin none # No spin specified, why?
partition_type rho_r2 # Patition type for integration grids
occupation_type gaussian 0.000001 # Broadening scheme used to find Fermi level
empty_states 25000 # Kohn-Sham states beyond the occupied levels
density_update_method density_matrix # Density matrix updating scheme
sc_accuracy_rho 1E-5 # SCF options
sc_accuracy_eev 1E-5
sc_accuracy_etot 1E-5
sc_iter_limit 400
#--------------------------------------------------------------
#-------------------GW specific options------------------------
anacon_type 1 # Pade analytic continuation
n_anacon_par 16 # Number of parameters in the Pade approximation
frequency_points 200 # Number of imaginary frequency points in \Sigma
#contour_def_gw 5 5 # Range of states for which CD is applied
#contour_eta 0.001 # Infinitesimal in Lehmann representation
state_lower_limit 1 # Lowest single-particle eigenstate
#--------------------------------------------------------------
#-------------------Relativistic options-----------------------
relativistic none # Non-relativistic calculation
override_relativity .true. # Do not stop the code when questionable input
#--------------------------------------------------------------
#-------------------Density mixing options---------------------
mixer pulay
n_max_pulay 10
charge_mix_param 0.2
#--------------------------------------------------------------
################################################################################
#
# FHI-aims code project
# VB, Fritz-Haber Institut, 2007
#
# Suggested "safe" defaults for H atom (to be pasted into control.in file)
#
################################################################################
species H
include_min_basis false
pure_gauss true
cut_pot 6.0 2.5 1.0
l_hartree 8
basis_dep_cutoff 0.d0
radial_base 100 7.0
radial_multiplier 8
angular_grids auto
angular 1202
angular_acc 1.0e-08
angular_min 110
basis_acc 1.0e-5
# global species definitions
nucleus 1
mass 1.00794
#
#
#
################################################################################
#
# Definition of "minimal" basis
#
################################################################################
# valence basis states
valence 1 s 1.
# ion occupancy
ion_occ 1 s 0.5
################################################################################
#
# Suggested additional basis functions. For production calculations,
# uncomment them one after another (the most important basis functions are
# listed first).
#
# Basis constructed for dimers: 0.5 A, 0.7 A, 1.0 A, 1.5 A, 2.5 A
#
################################################################################
# H cc-pVQZ
gaussian 0 3
82.6400000 0.0020060
12.4100000 0.0153430
2.8240000 0.0755790
gaussian 0 1 0.7977000
gaussian 0 1 0.2581000
gaussian 0 1 0.0898900
gaussian 1 1 2.2920000
gaussian 1 1 0.8380000
gaussian 1 1 0.2920000
gaussian 2 1 2.0620000
gaussian 2 1 0.6620000
gaussian 3 1 1.3970000
################################################################################
#
# FHI-aims code project
# VB, Fritz-Haber Institut, 2007
#
# Suggested "safe" defaults for O atom (to be pasted into control.in file)
#
################################################################################
species O
include_min_basis false
pure_gauss true
cut_pot 6.0 2.5 1.0
l_hartree 8
basis_dep_cutoff 0.d0
radial_base 100 7.0
radial_multiplier 8
angular_grids auto
angular 1202
angular_acc 1.0e-08
angular_min 110
basis_acc 1.0e-5
# global species definitions
nucleus 8
mass 15.9994
#
#
#
################################################################################
#
# Definition of "minimal" basis
#
################################################################################
# valence basis states
valence 2 s 2.
valence 2 p 4.
# ion occupancy
ion_occ 2 s 1.
ion_occ 2 p 3.
################################################################################
#
# Suggested additional basis functions. For production calculations,
# uncomment them one after another (the most important basis functions are
# listed first).
#
# Constructed for dimers: 1.0 A, 1.208 A, 1.5 A, 2.0 A, 3.0 A
#
################################################################################
# O cc-pVQZ
gaussian 0 9
61420.0000000 0.0000900
9199.0000000 0.0006980
2091.0000000 0.0036640
590.9000000 0.0152180
192.3000000 0.0524230
69.3200000 0.1459210
26.9700000 0.3052580
11.1000000 0.3985080
4.6820000 0.2169800
gaussian 0 9
61420.0000000 -0.0000200
9199.0000000 -0.0001590
2091.0000000 -0.0008290
590.9000000 -0.0035080
192.3000000 -0.0121560
69.3200000 -0.0362610
26.9700000 -0.0829920
11.1000000 -0.1520900
4.6820000 -0.1153310
gaussian 0 1 1.4280000
gaussian 0 1 0.5547000
gaussian 0 1 0.2067000
gaussian 1 3
63.4200000 0.0060440
14.6600000 0.0417990
4.4590000 0.1611430
gaussian 1 1 1.5310000
gaussian 1 1 0.5302000
gaussian 1 1 0.1750000
gaussian 2 1 3.7750000
gaussian 2 1 1.3000000
gaussian 2 1 0.4440000
gaussian 3 1 2.6660000
gaussian 3 1 0.8590000
gaussian 4 1 1.8460000
-----------------------------------------------------------------------
Completed first pass over input file control.in .
-----------------------------------------------------------------------
-----------------------------------------------------------------------
Parsing geometry.in (first pass over file, find array dimensions only).
The contents of geometry.in will be repeated verbatim below
unless switched off by setting 'verbatim_writeout .false.' .
in the first line of geometry.in .
-----------------------------------------------------------------------
atom 0.00000000 -0.00000000 -0.00614048 O
atom 0.76443318 -0.00000000 0.58917024 H
atom -0.76443318 0.00000000 0.58917024 H
-----------------------------------------------------------------------
Completed first pass over input file geometry.in .
-----------------------------------------------------------------------
Basic array size parameters:
| Number of species : 2
| Number of atoms : 3
| Max. basis fn. angular momentum : 4
| Max. atomic/ionic basis occupied n: 2
| Max. number of basis fn. types : 1
| Max. radial fns per species/type : 22
| Max. logarithmic grid size : 1430
| Max. radial integration grid size : 807
| Max. angular integration grid size: 1202
| Max. angular grid division number : 8
| Radial grid for Hartree potential : 1430
| Number of spin channels : 1
------------------------------------------------------------
Reading file control.in.
------------------------------------------------------------
XC: Using PBE gradient-corrected functionals.
GW quasiparticle calculation of excited states will be started after the DFT/HF calculation.
Kohn-Sham eigenvalues and eigenfunctions calculated by LAPACK via ELSI.
override_illconditioning: Explicitly overriding any built-in checks for an ill-conditioned overlap matrix.
*** WARNING: If you use this flag, you should really know what you are doing.
*** DO NOT keep this flag set by default in all your control.in files.
The 'V' version of RI (resolution of identity) technique is used.
Threshold for auxiliary basis singularities: 0.1000E-04
Spin treatment: No spin polarisation.
Partition function in integrals calculations: rho / r^2
Occupation type: Gaussian broadening, width = 0.100000E-05 eV.
Number of empty states per atom: 25000
Convergence accuracy of self-consistent charge density: 0.1000E-04
Convergence accuracy of sum of eigenvalues: 0.1000E-04
Convergence accuracy of total energy: 0.1000E-04
Maximum number of s.-c. iterations : 400
Using Pade approximation for analytical continuation.
Number of fitting parameters for analytical continuation : 16
Number of frequency points used for the self-energy calculation: 200
Lower limit of the eigenstates for the self-energy correction : 1
Non-relativistic treatment of kinetic energy.
override_relativity: Explicitly overriding any built-in relativity checks.
If you use this flag, you should really know what you are doing.
Using pulay charge density mixing.
Pulay mixing - number of memorized iterations: 10
Charge density mixing - mixing parameter: 0.2000
Reading configuration options for species H .
| Found request to include minimal basis fns. : F
| Found request to include pure gaussian fns. : T
| Found cutoff potl. onset [A], width [A], scale factor : 6.00000 2.50000 1.00000
| Found l_max for Hartree potential : 8
| Threshold for basis-dependent cutoff potential is 0.000000E+00
| Found data for basic radial integration grid : 100 points, outermost radius = 7.000 A
| Found multiplier for basic radial grid : 8
| Found angular grid specification: automatic.
| Found max. number of angular integration points per radial shell : 1202
| Found accuracy criterion for angular integrations : 0.1000E-07
| Will adapt angular grid densities automatically.
| Found min. number of angular integration points per radial shell : 110
| Found basis singularity cutoff : 0.1000E-04
| Found nuclear charge : 1.0000
| Found atomic mass : 1.00794000000000 amu
| Found free-atom valence shell : 1 s 1.000
| No ionic wave fns used. Skipping ion_occ.
| Found contracted cartesian Gaussian basis function : L = 0 , 3 elementary Gaussians:
| alpha = 0.826400E+02 weight = 0.200600E-02
| alpha = 0.124100E+02 weight = 0.153430E-01
| alpha = 0.282400E+01 weight = 0.755790E-01
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found primitive cartesian Gaussian basis function : 0 0.797700E+00
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found primitive cartesian Gaussian basis function : 0 0.258100E+00
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found primitive cartesian Gaussian basis function : 0 0.898900E-01
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found primitive cartesian Gaussian basis function : 1 0.229200E+01
| In terms of angular momentum, this radial function adds:
| 3 p-type basis functions
| Found primitive cartesian Gaussian basis function : 1 0.838000E+00
| In terms of angular momentum, this radial function adds:
| 3 p-type basis functions
| Found primitive cartesian Gaussian basis function : 1 0.292000E+00
| In terms of angular momentum, this radial function adds:
| 3 p-type basis functions
| Found primitive cartesian Gaussian basis function : 2 0.206200E+01
| In terms of angular momentum, this radial function adds:
| 5 d-type basis functions
| Found primitive cartesian Gaussian basis function : 2 0.662000E+00
| In terms of angular momentum, this radial function adds:
| 5 d-type basis functions
| Found primitive cartesian Gaussian basis function : 3 0.139700E+01
| In terms of angular momentum, this radial function adds:
| 7 f-type basis functions
Species H : Missing cutoff potential type.
Defaulting to exp(1/x)/(1-x)^2 type cutoff potential.
Species H : No 'logarithmic' tag. Using default grid for free atom:
| Default logarithmic grid data [bohr] : 0.1000E-03 0.1000E+03 0.1012E+01
Species H : Using default innermost maximum threshold i_radial= 2 for radial functions.
Species H : Default cutoff onset for free atom density etc. is infinite
since the product basis is used (hybrid functionals, Hartree-Fock, GW etc.).
Species H : Basic radial grid will be enhanced according to radial_multiplier = 8, to contain 807 grid points.
Reading configuration options for species O .
| Found request to include minimal basis fns. : F
| Found request to include pure gaussian fns. : T
| Found cutoff potl. onset [A], width [A], scale factor : 6.00000 2.50000 1.00000
| Found l_max for Hartree potential : 8
| Threshold for basis-dependent cutoff potential is 0.000000E+00
| Found data for basic radial integration grid : 100 points, outermost radius = 7.000 A
| Found multiplier for basic radial grid : 8
| Found angular grid specification: automatic.
| Found max. number of angular integration points per radial shell : 1202
| Found accuracy criterion for angular integrations : 0.1000E-07
| Will adapt angular grid densities automatically.
| Found min. number of angular integration points per radial shell : 110
| Found basis singularity cutoff : 0.1000E-04
| Found nuclear charge : 8.0000
| Found atomic mass : 15.9994000000000 amu
| Found free-atom valence shell : 2 s 2.000
| Found free-atom valence shell : 2 p 4.000
| No ionic wave fns used. Skipping ion_occ.
| No ionic wave fns used. Skipping ion_occ.
| Found contracted cartesian Gaussian basis function : L = 0 , 9 elementary Gaussians:
| alpha = 0.614200E+05 weight = 0.900000E-04
| alpha = 0.919900E+04 weight = 0.698000E-03
| alpha = 0.209100E+04 weight = 0.366400E-02
| alpha = 0.590900E+03 weight = 0.152180E-01
| alpha = 0.192300E+03 weight = 0.524230E-01
| alpha = 0.693200E+02 weight = 0.145921E+00
| alpha = 0.269700E+02 weight = 0.305258E+00
| alpha = 0.111000E+02 weight = 0.398508E+00
| alpha = 0.468200E+01 weight = 0.216980E+00
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found contracted cartesian Gaussian basis function : L = 0 , 9 elementary Gaussians:
| alpha = 0.614200E+05 weight = -.200000E-04
| alpha = 0.919900E+04 weight = -.159000E-03
| alpha = 0.209100E+04 weight = -.829000E-03
| alpha = 0.590900E+03 weight = -.350800E-02
| alpha = 0.192300E+03 weight = -.121560E-01
| alpha = 0.693200E+02 weight = -.362610E-01
| alpha = 0.269700E+02 weight = -.829920E-01
| alpha = 0.111000E+02 weight = -.152090E+00
| alpha = 0.468200E+01 weight = -.115331E+00
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found primitive cartesian Gaussian basis function : 0 0.142800E+01
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found primitive cartesian Gaussian basis function : 0 0.554700E+00
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found primitive cartesian Gaussian basis function : 0 0.206700E+00
| In terms of angular momentum, this radial function adds:
| 1 s-type basis function
| Found contracted cartesian Gaussian basis function : L = 1 , 3 elementary Gaussians:
| alpha = 0.634200E+02 weight = 0.604400E-02
| alpha = 0.146600E+02 weight = 0.417990E-01
| alpha = 0.445900E+01 weight = 0.161143E+00
| In terms of angular momentum, this radial function adds:
| 3 p-type basis functions
| Found primitive cartesian Gaussian basis function : 1 0.153100E+01
| In terms of angular momentum, this radial function adds:
| 3 p-type basis functions
| Found primitive cartesian Gaussian basis function : 1 0.530200E+00
| In terms of angular momentum, this radial function adds:
| 3 p-type basis functions
| Found primitive cartesian Gaussian basis function : 1 0.175000E+00
| In terms of angular momentum, this radial function adds:
| 3 p-type basis functions
| Found primitive cartesian Gaussian basis function : 2 0.377500E+01
| In terms of angular momentum, this radial function adds:
| 5 d-type basis functions
| Found primitive cartesian Gaussian basis function : 2 0.130000E+01
| In terms of angular momentum, this radial function adds:
| 5 d-type basis functions
| Found primitive cartesian Gaussian basis function : 2 0.444000E+00
| In terms of angular momentum, this radial function adds:
| 5 d-type basis functions
| Found primitive cartesian Gaussian basis function : 3 0.266600E+01
| In terms of angular momentum, this radial function adds:
| 7 f-type basis functions
| Found primitive cartesian Gaussian basis function : 3 0.859000E+00
| In terms of angular momentum, this radial function adds:
| 7 f-type basis functions
| Found primitive cartesian Gaussian basis function : 4 0.184600E+01
| In terms of angular momentum, this radial function adds:
| 9 g-type basis functions
Species O : Missing cutoff potential type.
Defaulting to exp(1/x)/(1-x)^2 type cutoff potential.
Species O : No 'logarithmic' tag. Using default grid for free atom:
| Default logarithmic grid data [bohr] : 0.1000E-03 0.1000E+03 0.1012E+01
Species O : Using default innermost maximum threshold i_radial= 2 for radial functions.
Species O : Default cutoff onset for free atom density etc. is infinite
since the product basis is used (hybrid functionals, Hartree-Fock, GW etc.).
Species O : Basic radial grid will be enhanced according to radial_multiplier = 8, to contain 807 grid points.
Finished reading input file 'control.in'.
------------------------------------------------------------
------------------------------------------------------------
Reading geometry description geometry.in.
------------------------------------------------------------
| The smallest distance between any two atoms is 0.96889264 AA.
| The first atom of this pair is atom number 1 .
| The second atom of this pair is atom number 2 .
Input structure read successfully.
The structure contains 3 atoms, and a total of 10.000 electrons.
Input geometry:
| No unit cell requested.
| Atomic structure:
| Atom x [A] y [A] z [A]
| 1: Species O 0.00000000 0.00000000 -0.00614048
| 2: Species H 0.76443318 0.00000000 0.58917024
| 3: Species H -0.76443318 0.00000000 0.58917024
Finished reading input file 'control.in'.
------------------------------------------------------------
Reading geometry description geometry.in.
------------------------------------------------------------
Consistency checks for stacksize environment parameter are next.
| Maximum stacksize for task 0: unlimited
| Maximum stacksize for task 1: unlimited
| Maximum stacksize for task 2: unlimited
| Maximum stacksize for task 3: unlimited
| Maximum stacksize for task 4: unlimited
| Maximum stacksize for task 5: unlimited
| Maximum stacksize for task 6: unlimited
| Maximum stacksize for task 7: unlimited
| Current stacksize for task 0: unlimited
| Current stacksize for task 1: unlimited
| Current stacksize for task 2: unlimited
| Current stacksize for task 3: unlimited
| Current stacksize for task 4: unlimited
| Current stacksize for task 5: unlimited
| Current stacksize for task 6: unlimited
| Current stacksize for task 7: unlimited
Consistency checks for the contents of control.in are next.
MPI_IN_PLACE appears to work with this MPI implementation.
| Keeping use_mpi_in_place .true. (see manual).
Species H: Using default value for prodbas_acc = 1.000000E-02.
Species H: Using default value max_l_prodbas = 5.
Species O: Using default value for prodbas_acc = 1.000000E-02.
Species O: Using default value max_l_prodbas = 5.
* Species O: Specified min. number of angular integration points is 110
* The angular momenta for this species require 194 for RI_type 'V'. Increasing angular_min to 194.
Target number of points in a grid batch is not set. Defaulting to 100
Method for grid partitioning is not set. Defaulting to parallel hash+maxmin partitioning.
Batch size limit is not set. Defaulting to 200
By default, will store active basis functions for each batch.
If in need of memory, prune_basis_once .false. can be used to disable this option.
communication_type for Hartree potential was not specified.
Defaulting to calc_hartree .
Pulay mixer: Number of initial linear mixing iterations not set.
Defaulting to 0 iterations.
Work space size for distributed Hartree potential not set.
Defaulting to 0.200000E+03 MB.
Algorithm-dependent basis array size parameters:
| n_max_pulay : 10
Presetting 1001 iterations before the initial mixing cycle
is restarted anyway using the sc_init_iter criterion / keyword.
Presetting a factor 1.000 between actual scf density residual
and density convergence criterion sc_accuracy_rho below which sc_init_iter
takes no effect.
Calculation of forces was not defined in control.in. No forces will be calculated.
Geometry relaxation not requested: no relaxation will be performed.
No accuracy limit for integral partition fn. given. Defaulting to 0.1000E-14.
No threshold value for u(r) in integrations given. Defaulting to 0.1000E-05.
No accuracy for occupation numbers given. Defaulting to 0.1000E-12.
No threshold value for occupation numbers given. Defaulting to 0.0000E+00.
No accuracy for fermi level given. Defaulting to 0.1000E-19.
Maximum # of iterations to find E_F not set. Defaulting to 200.
Will not use alltoall communication since running on < 1024 CPUs.
Threshold for basis singularities not set.
Default threshold for basis singularities: 0.1000E-04
Partitioning for Hartree potential was not defined. Using partition_type for integrals.
| Reporting present value of keyword multip_moments_threshold : 0.10000000E-09
| This value may affect high angular momentum components of the Hartree potential in periodic systems.
* Doing correlated calculations, so all empty single-particle states will be included.
No q(lm)/r^(l+1) cutoff set for long-range Hartree potential.
| Using default value of 0.100000E-09 .
| Verify using the multipole_threshold keyword.
Defaulting to new monopole extrapolation.
Density update method: density matrix based density update selected.
Charge integration errors on the 3D integration grid will be compensated
by explicit normalization and distribution of residual charges.
Use the "compensate_multipole_errors" flag to change this behaviour.
Default to 1D ("use_logsbt") integrations for auxiliary 2-center integrals.
Default onset of logarithmic r-grid for SBT is -38.000000000000
Default onset of logarithmic k-grid for SBT is -25.000000000000
Default range of logarithmic r- and k-grid for SBT is 45.000000000000
Default number of logarithmic r- and k-grid for SBT is 4096
Set 'collect_eigenvectors' to be '.true.' for all serial calculations. This is mandatory.
Set 'collect_eigenvectors' to be '.true.' for GW/RPA/MP2, SCREX/COHSEX cluster calculations
Set 'collect_eigenvectors' to be '.true.' for qpe_calc (quasiparticle energy).
Set 'collect_eigenvectors' to be '.true.' for KS_method lapack_fast and serial.
Consistency checks for the contents of geometry.in are next.
Number of empty states per atom not set in control.in .
| Since you are using a method that relies on the unoccupied spectrum
| (MP2,GW,RPA et al.), will use the full Hamiltonian size (see below)
| as the max. possible number of states (occupied plus empty).
Structure-dependent array size parameters:
| Maximum number of distinct radial functions : 25
| Maximum number of basis functions : 115
| Number of Kohn-Sham states (occupied + empty): 115
------------------------------------------------------------
------------------------------------------------------------
Preparing all fixed parts of the calculation.
------------------------------------------------------------
Determining machine precision:
2.225073858507201E-308
Setting up grids for atomic and cluster calculations.
* Minimum radial grid point for logarithmic grid of species 1
* , r_min = 9.999999747378752E-005 ,
* is chosen above the minimum radial integration grid point,
* r_min = 5.159655215165401E-006 .
* Setting r_grid_min to 2.579827607582700E-006 .
* Minimum radial grid point for logarithmic grid of species 2
* , r_min = 1.249999968422344E-005 ,
* is chosen above the minimum radial integration grid point,
* r_min = 2.063866827914613E-005 .
* Setting r_grid_min to 2.579827607582700E-006 .
Creating wave function, potential, and density for free atoms.
Species: H
List of occupied orbitals and eigenvalues:
n l occ energy [Ha] energy [eV]
1 0 1.0000 -0.238600 -6.4926
Species: O
List of occupied orbitals and eigenvalues:
n l occ energy [Ha] energy [eV]
1 0 2.0000 -18.898644 -514.2583
2 0 2.0000 -0.878848 -23.9147
2 1 4.0000 -0.332128 -9.0377
Adding cutoff potential to free-atom effective potential.
Creating fixed part of basis set: Ionic, confined, hydrogenic.
H Gaussian:
List of cartesian Gaussian basis orbitals:
L l
0 0
0 0
0 0
0 0
1 1
1 1
1 1
2 2
2 2
3 3
O Gaussian:
List of cartesian Gaussian basis orbitals:
L l
0 0
0 0
0 0
0 0
0 0
1 1
1 1
1 1
1 1
2 2
2 2
2 2
3 3
3 3
4 4
Creating atomic-like basis functions for current effective potential.
Assembling full basis from fixed parts.
| Species H : gaussian orbital 0 s accepted.
| Species H : gaussian orbital 0 s accepted.
| Species H : gaussian orbital 0 s accepted.
| Species H : gaussian orbital 0 s accepted.
| Species H : gaussian orbital 1 p accepted.
| Species H : gaussian orbital 1 p accepted.
| Species H : gaussian orbital 1 p accepted.
| Species H : gaussian orbital 2 d accepted.
| Species H : gaussian orbital 2 d accepted.
| Species H : gaussian orbital 3 f accepted.
| Species O : gaussian orbital 0 s accepted.
| Species O : gaussian orbital 0 s accepted.
| Species O : gaussian orbital 0 s accepted.
| Species O : gaussian orbital 0 s accepted.
| Species O : gaussian orbital 0 s accepted.
| Species O : gaussian orbital 1 p accepted.
| Species O : gaussian orbital 1 p accepted.
| Species O : gaussian orbital 1 p accepted.
| Species O : gaussian orbital 1 p accepted.
| Species O : gaussian orbital 2 d accepted.
| Species O : gaussian orbital 2 d accepted.
| Species O : gaussian orbital 2 d accepted.
| Species O : gaussian orbital 3 f accepted.
| Species O : gaussian orbital 3 f accepted.
| Species O : gaussian orbital 4 g accepted.
Basis size parameters after reduction:
| Total number of radial functions: 25
| Total number of basis functions : 115
Per-task memory consumption for arrays in subroutine allocate_ext:
| 6.868128MB.
Testing on-site integration grid accuracy.
| Species Function <phi|h_atom|phi> (log., in eV) <phi|h_atom|phi> (rad., in eV)
1 1 66.9543016856 66.9543016814
1 2 80.9440492330 80.9440491616
1 3 46.2485412815 46.2485410915
1 4 22.9542283999 22.9542281783
1 5 119.9150794209 119.9150792718
1 6 95.4140689984 95.4140683682
1 7 50.4865898892 50.4865891091
1 8 171.0666911560 171.0666907483
1 9 87.9142398331 87.9142390949
1 10 155.3029127732 155.3029121495
2 11 -514.0595839405 -514.0595854029
2 12 607.0111451249 607.0111405413
2 13 274.6072326348 274.6072316254
2 14 153.5325979405 153.5325971029
2 15 73.7225913875 73.7225905602
2 16 81.3565108203 81.3565107901
2 17 122.5303011331 122.5303007681
2 18 75.8746728709 75.8746721168
2 19 34.3363428878 34.3363422739
2 20 218.9177453542 218.9177447653
2 21 147.8581850352 147.8581835597
2 22 71.7831007132 71.7830993479
2 23 249.7504167368 249.7504156988
2 24 120.9441017010 120.9441003027
2 25 235.2477364776 235.2477350153
Preparing densities etc. for the partition functions (integrals / Hartree potential).
Preparations completed.
max(cpu_time) : 0.372 s.
Wall clock time (cpu1) : 2.049 s.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency loop: Initialization.
Date : 20210920, Time : 144852.110
------------------------------------------------------------
Initializing index lists of integration centers etc. from given atomic structure:
| Number of centers in hartree potential : 3
| Number of centers in hartree multipole : 3
| Number of centers in electron density summation: 3
| Number of centers in basis integrals : 3
| Number of centers in integrals : 3
| Number of centers in hamiltonian : 3
Allocating 0.101 MB for KS_eigenvector
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 0
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 1
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 2
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 3
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 4
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 5
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 6
| Estimated number of non-zero basis functions for the Hamiltonian : 115 in task 7
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 0
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 1
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 2
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 3
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 4
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 5
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 6
| Estimated number of non-zero radial functions for the Hamiltonian: 35 in task 7
Initial 3D integrations: Overlap and Hamiltonian matrix.
| Adapting angular integration grids if requested.
Output of integration grids in suitable form for copy-paste into control.in:
Species H :
division 0.3258 110
division 0.4845 194
division 0.6174 302
division 0.6732 434
division 0.7063 590
division 0.7407 770
division 0.7604 974
division 1.1972 1202
division 1.3045 974
division 1.6873 1202
division 1.8983 974
division 2.0980 770
division 2.2615 590
division 2.5703 434
division 2.7609 302
division 3.1562 194
outer_grid 110
Species O :
division 0.4596 194
division 0.5651 302
division 0.6310 434
division 0.6841 590
division 0.7446 770
division 0.7724 974
division 1.1674 1202
division 1.2340 974
division 1.3245 770
division 1.5929 590
division 2.0170 434
division 2.2352 302
outer_grid 194
Partitioning the integration grid into batches with parallel hashing+maxmin method.
| Number of batches: 13146
| Maximal batch size: 199
| Minimal batch size: 49
| Average batch size: 74.812
| Standard deviation of batch sizes: 24.349
Integration load balanced across 8 MPI tasks.
Work distribution over tasks is as follows:
Task 0 has 122903 integration points.
Task 1 has 122966 integration points.
Task 2 has 122966 integration points.
Task 3 has 122970 integration points.
Task 4 has 122932 integration points.
Task 5 has 122906 integration points.
Task 6 has 122930 integration points.
Task 7 has 122905 integration points.
Initializing partition tables, free-atom densities, potentials, etc. across the integration grid (initialize_grid_storage).
| Net number of integration points: 983478
| of which are non-zero points : 983105
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 9.9999999715
| Charge integration error : -0.0000000285
| Normalization factor for density and gradient : 1.0000000028
Renormalizing the free-atom superposition density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 9.9999999715
| Charge integration error : -0.0000000285
| Normalization factor for density and gradient : 1.0000000028
Obtaining max. number of non-zero basis functions in each batch (get_n_compute_maxes).
| Maximal number of non-zero basis functions: 115 in task 0
| Maximal number of non-zero basis functions: 115 in task 1
| Maximal number of non-zero basis functions: 115 in task 2
| Maximal number of non-zero basis functions: 115 in task 3
| Maximal number of non-zero basis functions: 115 in task 4
| Maximal number of non-zero basis functions: 115 in task 5
| Maximal number of non-zero basis functions: 115 in task 6
| Maximal number of non-zero basis functions: 115 in task 7
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Overlap matrix is not singular
| Lowest and highest eigenvalues : 0.2953E-03, 0.2968E+01
Finished singularity check of overlap matrix
| Time : 0.202 s
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.003 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -10.21158211 eV
Writing Kohn-Sham eigenvalues.
State Occupation Eigenvalue [Ha] Eigenvalue [eV]
1 2.00000 -19.005579 -517.16811
2 2.00000 -1.097557 -29.86604
3 2.00000 -0.628537 -17.10337
4 2.00000 -0.510442 -13.88984
5 2.00000 -0.437691 -11.91017
6 0.00000 -0.055997 -1.52377
7 0.00000 0.011523 0.31357
8 0.00000 0.208102 5.66275
9 0.00000 0.233422 6.35172
10 0.00000 0.251987 6.85692
11 0.00000 0.305461 8.31201
12 0.00000 0.370820 10.09051
13 0.00000 0.377209 10.26437
14 0.00000 0.424630 11.55477
15 0.00000 0.485386 13.20803
16 0.00000 0.561149 15.26964
17 0.00000 0.704608 19.17335
18 0.00000 0.830510 22.59934
19 0.00000 0.865633 23.55506
20 0.00000 1.066711 29.02669
21 0.00000 1.100337 29.94170
22 0.00000 1.124536 30.60019
23 0.00000 1.128950 30.72029
24 0.00000 1.217001 33.11627
25 0.00000 1.280594 34.84674
26 0.00000 1.494005 40.65394
27 0.00000 1.715386 46.67804
28 0.00000 1.831143 49.82794
29 0.00000 1.925341 52.39120
30 0.00000 1.942090 52.84696
31 0.00000 2.042507 55.57945
32 0.00000 2.141745 58.27984
33 0.00000 2.156300 58.67590
34 0.00000 2.163233 58.86458
35 0.00000 2.242771 61.02890
36 0.00000 2.277364 61.97023
37 0.00000 2.307687 62.79537
38 0.00000 2.382020 64.81805
39 0.00000 2.465521 67.09024
40 0.00000 2.569134 69.90969
41 0.00000 2.697074 73.39112
42 0.00000 2.850768 77.57335
43 0.00000 2.851716 77.59913
44 0.00000 2.956074 80.43885
45 0.00000 3.124010 85.00863
46 0.00000 3.258554 88.66977
47 0.00000 3.422981 93.14405
48 0.00000 3.514278 95.62836
49 0.00000 3.589964 97.68790
50 0.00000 3.624287 98.62186
51 0.00000 3.877014 105.49891
52 0.00000 3.879192 105.55819
53 0.00000 4.085937 111.18399
54 0.00000 4.150347 112.93669
55 0.00000 4.240638 115.39364
56 0.00000 4.318973 117.52524
57 0.00000 4.726664 128.61908
58 0.00000 4.801792 130.66342
59 0.00000 5.506839 149.84872
60 0.00000 5.604102 152.49537
61 0.00000 5.770314 157.01824
62 0.00000 5.842305 158.97722
63 0.00000 6.155418 167.49746
64 0.00000 6.247669 170.00772
65 0.00000 6.483759 176.43205
66 0.00000 6.656734 181.13896
67 0.00000 6.745036 183.54177
68 0.00000 6.824300 185.69864
69 0.00000 6.886585 187.39353
70 0.00000 6.929565 188.56305
71 0.00000 7.034278 191.41244
72 0.00000 7.066609 192.29221
73 0.00000 7.465569 203.14848
74 0.00000 7.480478 203.55418
75 0.00000 7.544405 205.29371
76 0.00000 7.571692 206.03623
77 0.00000 7.617645 207.28666
78 0.00000 7.718813 210.03958
79 0.00000 7.727304 210.27065
80 0.00000 7.812416 212.58667
81 0.00000 7.852324 213.67260
82 0.00000 8.061942 219.37661
83 0.00000 8.178506 222.54847
84 0.00000 8.265488 224.91536
85 0.00000 8.270171 225.04281
86 0.00000 8.557463 232.86041
87 0.00000 8.642360 235.17059
88 0.00000 8.801426 239.49900
89 0.00000 9.118121 248.11669
90 0.00000 9.239841 251.42886
91 0.00000 9.451911 257.19959
92 0.00000 9.609562 261.48949
93 0.00000 9.635719 262.20125
94 0.00000 9.911668 269.71021
95 0.00000 9.957202 270.94925
96 0.00000 10.426230 283.71215
97 0.00000 10.546566 286.98667
98 0.00000 10.629426 289.24141
99 0.00000 10.745092 292.38882
100 0.00000 10.758110 292.74308
101 0.00000 10.912691 296.94944
102 0.00000 11.252359 306.19225
103 0.00000 11.322971 308.11371
104 0.00000 11.448627 311.53299
105 0.00000 11.463578 311.93982
106 0.00000 11.558069 314.51107
107 0.00000 12.623675 343.50769
108 0.00000 12.877868 350.42462
109 0.00000 13.285474 361.51614
110 0.00000 13.629071 370.86589
111 0.00000 13.664729 371.83621
112 0.00000 13.771876 374.75182
113 0.00000 15.434693 419.99937
114 0.00000 15.787416 429.59744
115 0.00000 42.140054 1146.68920
Highest occupied state (VBM) at -11.91017390 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -1.52376726 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 10.38640664 eV.
Calculating total energy contributions from superposition of free atom densities.
Total energy components:
| Sum of eigenvalues : -43.35961131 Ha -1179.87505513 eV
| XC energy correction : -9.01212505 Ha -245.23239984 eV
| XC potential correction : 11.58761901 Ha 315.31515619 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : 0.00000000 Ha 0.00000000 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.46025746 Ha -2080.58946463 eV
| Total energy, T -> 0 : -76.46025746 Ha -2080.58946463 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.46025746 Ha -2080.58946463 eV
Derived energy quantities:
| Kinetic energy : 75.52297145 Ha 2055.08461470 eV
| Electrostatic energy : -142.97110386 Ha -3890.44167950 eV
| Energy correction for multipole
| error in Hartree potential : 0.00000000 Ha 0.00000000 eV
| Sum of eigenvalues per atom : -393.29168504 eV
| Total energy (T->0) per atom : -693.52982154 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -693.52982154 eV
Initialize hartree_potential_storage
Max. number of atoms included in rho_multipole: 3
End scf initialization - timings : max(cpu_time) wall_clock(cpu1)
| Time for scf. initialization : 5.520 s 5.896 s
| Boundary condition initialization : 0.000 s 0.001 s
| Integration : 4.604 s 4.609 s
| Solution of K.-S. eqns. : 0.008 s 0.209 s
| Grid partitioning : 0.244 s 0.243 s
| Preloading free-atom quantities on grid : 0.220 s 0.276 s
| Free-atom superposition energy : 0.068 s 0.066 s
| Total energy evaluation : 0.000 s 0.001 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.204 MB (on task 0)
| Maximum: 0.204 MB (on task 0)
| Average: 0.204 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.804 s, elapsed 3.815 s
Integration grid: deviation in total charge (<rho> - N_e) = 9.702012E-09
Time for density update prior : max(cpu_time) wall_clock(cpu1)
| self-consistency iterative process : 0.496 s 0.540 s
------------------------------------------------------------
Begin self-consistency iteration # 1
Date : 20210920, Time : 144858.547
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000019
| Charge integration error : 0.0000000019
| Normalization factor for density and gradient : 0.9999999998
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.126789E-12
| Sum of charges compensated after spline to logarithmic grids = 0.111105E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.126728E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.908 s, elapsed 0.928 s
| RMS charge density error from multipole expansion : 0.276210E-03
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.937 s, elapsed 2.945 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -6.00875975 eV
Writing Kohn-Sham eigenvalues.
State Occupation Eigenvalue [Ha] Eigenvalue [eV]
1 2.00000 -18.845754 -512.81905
2 2.00000 -1.019707 -27.74764
3 2.00000 -0.561684 -15.28421
4 2.00000 -0.431475 -11.74102
5 2.00000 -0.355656 -9.67790
6 0.00000 -0.039230 -1.06751
7 0.00000 0.029865 0.81267
8 0.00000 0.226726 6.16953
9 0.00000 0.258915 7.04544
10 0.00000 0.270755 7.36762
11 0.00000 0.338480 9.21051
12 0.00000 0.394497 10.73481
13 0.00000 0.394891 10.74554
14 0.00000 0.440483 11.98614
15 0.00000 0.503195 13.69264
16 0.00000 0.592124 16.11251
17 0.00000 0.714911 19.45371
18 0.00000 0.850273 23.13711
19 0.00000 0.884627 24.07192
20 0.00000 1.089177 29.63801
21 0.00000 1.122803 30.55302
22 0.00000 1.159121 31.54130
23 0.00000 1.163909 31.67158
24 0.00000 1.254473 34.13595
25 0.00000 1.315330 35.79195
26 0.00000 1.530878 41.65730
27 0.00000 1.738538 47.30802
28 0.00000 1.854780 50.47114
29 0.00000 1.945732 52.94605
30 0.00000 1.995320 54.29542
31 0.00000 2.058231 56.00732
32 0.00000 2.164417 58.89680
33 0.00000 2.170730 59.06856
34 0.00000 2.189144 59.56964
35 0.00000 2.260376 61.50797
36 0.00000 2.300730 62.60604
37 0.00000 2.333781 63.50540
38 0.00000 2.426100 66.01755
39 0.00000 2.489672 67.74743
40 0.00000 2.607604 70.95652
41 0.00000 2.733028 74.36948
42 0.00000 2.883054 78.45190
43 0.00000 2.897386 78.84189
44 0.00000 2.996990 81.55225
45 0.00000 3.160849 86.01109
46 0.00000 3.315271 90.21312
47 0.00000 3.476536 94.60137
48 0.00000 3.580241 97.42331
49 0.00000 3.652712 99.39536
50 0.00000 3.683734 100.23950
51 0.00000 3.927482 106.87223
52 0.00000 3.936745 107.12429
53 0.00000 4.134236 112.49827
54 0.00000 4.200310 114.29626
55 0.00000 4.289298 116.71773
56 0.00000 4.369750 118.90694
57 0.00000 4.776123 129.96492
58 0.00000 4.847305 131.90189
59 0.00000 5.532578 150.54911
60 0.00000 5.632012 153.25485
61 0.00000 5.795456 157.70237
62 0.00000 5.865208 159.60044
63 0.00000 6.187897 168.38125
64 0.00000 6.280679 170.90598
65 0.00000 6.519541 177.40574
66 0.00000 6.698200 182.26729
67 0.00000 6.781276 184.52792
68 0.00000 6.856069 186.56312
69 0.00000 6.915657 188.18459
70 0.00000 6.968951 189.63479
71 0.00000 7.056939 192.02908
72 0.00000 7.090039 192.92977
73 0.00000 7.495051 203.95071
74 0.00000 7.504440 204.20619
75 0.00000 7.571210 206.02311
76 0.00000 7.600714 206.82594
77 0.00000 7.645874 208.05481
78 0.00000 7.750872 210.91197
79 0.00000 7.753702 210.98897
80 0.00000 7.832888 213.14371
81 0.00000 7.879392 214.40917
82 0.00000 8.088824 220.10810
83 0.00000 8.219162 223.65477
84 0.00000 8.305361 226.00038
85 0.00000 8.310378 226.13689
86 0.00000 8.597897 233.96069
87 0.00000 8.694244 236.58242
88 0.00000 8.831384 240.31419
89 0.00000 9.195339 250.21791
90 0.00000 9.310294 253.34600
91 0.00000 9.527247 259.24957
92 0.00000 9.705339 264.09570
93 0.00000 9.713249 264.31096
94 0.00000 9.986889 271.75708
95 0.00000 10.031900 272.98190
96 0.00000 10.499216 285.69821
97 0.00000 10.623793 289.08812
98 0.00000 10.725998 291.86926
99 0.00000 10.835616 294.85211
100 0.00000 10.846150 295.13876
101 0.00000 11.009205 299.57570
102 0.00000 11.333042 308.38775
103 0.00000 11.421767 310.80209
104 0.00000 11.545484 314.16860
105 0.00000 11.551464 314.33133
106 0.00000 11.645350 316.88608
107 0.00000 12.713886 345.96245
108 0.00000 12.947264 352.31299
109 0.00000 13.374306 363.93337
110 0.00000 13.719657 373.33087
111 0.00000 13.756311 374.32828
112 0.00000 13.865219 377.29180
113 0.00000 15.517552 422.25409
114 0.00000 15.873763 431.94706
115 0.00000 42.282241 1150.55831
Highest occupied state (VBM) at -9.67789747 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -1.06750974 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 8.61038772 eV.
Total energy components:
| Sum of eigenvalues : -42.42855168 Ha -1154.53963341 eV
| XC energy correction : -9.11899017 Ha -248.14034786 eV
| XC potential correction : 11.72764512 Ha 319.12546052 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -0.90758186 Ha -24.69655895 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.40361870 Ha -2079.04824556 eV
| Total energy, T -> 0 : -76.40361870 Ha -2079.04824556 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.40361870 Ha -2079.04824556 eV
Derived energy quantities:
| Kinetic energy : 76.13556931 Ha 2071.75425063 eV
| Electrostatic energy : -143.42019783 Ha -3902.66214834 eV
| Energy correction for multipole
| error in Hartree potential : 0.00001823 Ha 0.00049603 eV
| Sum of eigenvalues per atom : -384.84654447 eV
| Total energy (T->0) per atom : -693.01608185 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -693.01608185 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.709 s, elapsed 3.724 s
Integration grid: deviation in total charge (<rho> - N_e) = 9.713087E-09
Self-consistency convergence accuracy:
| Change of charge density : 0.3151E+00
| Change of sum of eigenvalues : 0.2534E+02 eV
| Change of total energy : 0.1541E+01 eV
------------------------------------------------------------
End self-consistency iteration # 1 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.012 s 1.011 s
| Charge density update : 0.476 s 0.473 s
| Density mixing : 0.028 s 0.026 s
| Hartree multipole update : 0.020 s 0.020 s
| Hartree multipole summation : 0.120 s 0.119 s
| Integration : 0.368 s 0.369 s
| Solution of K.-S. eqns. : 0.004 s 0.001 s
| Total energy evaluation : 0.004 s 0.001 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.204 MB (on task 0)
| Maximum: 0.204 MB (on task 0)
| Average: 0.204 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 2
Date : 20210920, Time : 144859.564
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000031
| Charge integration error : 0.0000000031
| Normalization factor for density and gradient : 0.9999999997
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.132065E-12
| Sum of charges compensated after spline to logarithmic grids = 0.121996E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.132336E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.915 s, elapsed 0.933 s
| RMS charge density error from multipole expansion : 0.653310E-03
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.939 s, elapsed 2.947 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.09736184 eV
Highest occupied state (VBM) at -7.91264589 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.64044686 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 7.27219904 eV.
Checking to see if s.c.f. parameters should be adjusted.
Total energy components:
| Sum of eigenvalues : -41.71948100 Ha -1135.24483854 eV
| XC energy correction : -9.20445217 Ha -250.46588720 eV
| XC potential correction : 11.83958729 Ha 322.17156201 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.62600774 Ha -44.24592184 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38649373 Ha -2078.58225144 eV
| Total energy, T -> 0 : -76.38649373 Ha -2078.58225144 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38649373 Ha -2078.58225144 eV
Derived energy quantities:
| Kinetic energy : 76.44210162 Ha 2080.09541928 eV
| Electrostatic energy : -143.62414318 Ha -3908.21178351 eV
| Energy correction for multipole
| error in Hartree potential : 0.00002364 Ha 0.00064317 eV
| Sum of eigenvalues per atom : -378.41494618 eV
| Total energy (T->0) per atom : -692.86075048 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.86075048 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.739 s, elapsed 3.761 s
Integration grid: deviation in total charge (<rho> - N_e) = 9.864300E-09
Self-consistency convergence accuracy:
| Change of charge density : 0.1715E+00
| Change of sum of eigenvalues : 0.1929E+02 eV
| Change of total energy : 0.4660E+00 eV
------------------------------------------------------------
End self-consistency iteration # 2 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.036 s 1.034 s
| Charge density update : 0.480 s 0.478 s
| Density mixing : 0.048 s 0.045 s
| Hartree multipole update : 0.020 s 0.020 s
| Hartree multipole summation : 0.120 s 0.120 s
| Integration : 0.372 s 0.369 s
| Solution of K.-S. eqns. : 0.004 s 0.001 s
| Total energy evaluation : 0.004 s 0.000 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 3
Date : 20210920, Time : 144900.598
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000083
| Charge integration error : 0.0000000083
| Normalization factor for density and gradient : 0.9999999992
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.161484E-12
| Sum of charges compensated after spline to logarithmic grids = 0.124247E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.161583E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.920 s, elapsed 0.936 s
| RMS charge density error from multipole expansion : 0.133168E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.938 s, elapsed 2.945 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.72158492 eV
Highest occupied state (VBM) at -7.26292315 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.27741147 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.98551168 eV.
Total energy components:
| Sum of eigenvalues : -41.57237379 Ha -1131.24184767 eV
| XC energy correction : -9.22680614 Ha -251.07416956 eV
| XC potential correction : 11.86863692 Ha 322.96204282 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.77683686 Ha -48.35019088 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38351996 Ha -2078.50133116 eV
| Total energy, T -> 0 : -76.38351996 Ha -2078.50133116 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38351996 Ha -2078.50133116 eV
Derived energy quantities:
| Kinetic energy : 76.05824754 Ha 2069.65021814 eV
| Electrostatic energy : -143.21496136 Ha -3897.07737974 eV
| Energy correction for multipole
| error in Hartree potential : -0.00001291 Ha -0.00035130 eV
| Sum of eigenvalues per atom : -377.08061589 eV
| Total energy (T->0) per atom : -692.83377705 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83377705 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.829 s, elapsed 3.852 s
Integration grid: deviation in total charge (<rho> - N_e) = 9.978617E-09
Self-consistency convergence accuracy:
| Change of charge density : 0.1073E+00
| Change of sum of eigenvalues : 0.4003E+01 eV
| Change of total energy : 0.8092E-01 eV
------------------------------------------------------------
End self-consistency iteration # 3 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.080 s 1.077 s
| Charge density update : 0.492 s 0.489 s
| Density mixing : 0.076 s 0.076 s
| Hartree multipole update : 0.020 s 0.020 s
| Hartree multipole summation : 0.124 s 0.121 s
| Integration : 0.368 s 0.368 s
| Solution of K.-S. eqns. : 0.004 s 0.002 s
| Total energy evaluation : 0.004 s 0.001 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 4
Date : 20210920, Time : 144901.675
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000039
| Charge integration error : 0.0000000039
| Normalization factor for density and gradient : 0.9999999996
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.551727E-13
| Sum of charges compensated after spline to logarithmic grids = 0.129635E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.548284E-13
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.915 s, elapsed 0.934 s
| RMS charge density error from multipole expansion : 0.146621E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.937 s, elapsed 2.946 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.15281373 eV
Highest occupied state (VBM) at -6.86433256 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.13358520 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.73074736 eV.
Total energy components:
| Sum of eigenvalues : -41.41738784 Ha -1127.02446540 eV
| XC energy correction : -9.24778429 Ha -251.64501422 eV
| XC potential correction : 11.89612026 Ha 323.70990254 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.93813406 Ha -52.73931111 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38332603 Ha -2078.49605406 eV
| Total energy, T -> 0 : -76.38332603 Ha -2078.49605406 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38332603 Ha -2078.49605406 eV
Derived energy quantities:
| Kinetic energy : 76.10046857 Ha 2070.79911089 eV
| Electrostatic energy : -143.23601031 Ha -3897.65015073 eV
| Energy correction for multipole
| error in Hartree potential : -0.00001755 Ha -0.00047750 eV
| Sum of eigenvalues per atom : -375.67482180 eV
| Total energy (T->0) per atom : -692.83201802 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83201802 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.828 s, elapsed 3.851 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.003486E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.2155E-01
| Change of sum of eigenvalues : 0.4217E+01 eV
| Change of total energy : 0.5277E-02 eV
------------------------------------------------------------
End self-consistency iteration # 4 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.076 s 1.076 s
| Charge density update : 0.492 s 0.489 s
| Density mixing : 0.076 s 0.076 s
| Hartree multipole update : 0.020 s 0.019 s
| Hartree multipole summation : 0.124 s 0.121 s
| Integration : 0.372 s 0.369 s
| Solution of K.-S. eqns. : 0.004 s 0.001 s
| Total energy evaluation : 0.004 s 0.000 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 5
Date : 20210920, Time : 144902.751
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000077
| Charge integration error : 0.0000000077
| Normalization factor for density and gradient : 0.9999999992
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.151670E-12
| Sum of charges compensated after spline to logarithmic grids = 0.130643E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.151276E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.919 s, elapsed 0.936 s
| RMS charge density error from multipole expansion : 0.145049E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.868 s, elapsed 2.888 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.26983142 eV
Highest occupied state (VBM) at -6.99120341 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.17198812 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81921529 eV.
Total energy components:
| Sum of eigenvalues : -41.46283698 Ha -1128.26119945 eV
| XC energy correction : -9.24177234 Ha -251.48142058 eV
| XC potential correction : 11.88821756 Ha 323.49485900 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89074619 Ha -51.44982149 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327805 Ha -2078.49474838 eV
| Total energy, T -> 0 : -76.38327805 Ha -2078.49474838 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327805 Ha -2078.49474838 eV
Derived energy quantities:
| Kinetic energy : 76.12085940 Ha 2071.35397371 eV
| Electrostatic energy : -143.26236511 Ha -3898.36730152 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002530 Ha -0.00068838 eV
| Sum of eigenvalues per atom : -376.08706648 eV
| Total energy (T->0) per atom : -692.83158279 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158279 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.828 s, elapsed 3.851 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001781E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.8436E-02
| Change of sum of eigenvalues : -0.1237E+01 eV
| Change of total energy : 0.1306E-02 eV
------------------------------------------------------------
End self-consistency iteration # 5 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.084 s 1.083 s
| Charge density update : 0.492 s 0.489 s
| Density mixing : 0.092 s 0.089 s
| Hartree multipole update : 0.020 s 0.020 s
| Hartree multipole summation : 0.124 s 0.121 s
| Integration : 0.364 s 0.361 s
| Solution of K.-S. eqns. : 0.004 s 0.002 s
| Total energy evaluation : 0.004 s 0.001 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 6
Date : 20210920, Time : 144903.835
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000027
| Charge integration error : 0.0000000027
| Normalization factor for density and gradient : 0.9999999997
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.887486E-13
| Sum of charges compensated after spline to logarithmic grids = 0.131579E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.886748E-13
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.916 s, elapsed 0.934 s
| RMS charge density error from multipole expansion : 0.145963E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.861 s, elapsed 2.872 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.001 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.24966244 eV
Highest occupied state (VBM) at -6.97465163 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16264362 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81200801 eV.
Total energy components:
| Sum of eigenvalues : -41.45656884 Ha -1128.09063473 eV
| XC energy correction : -9.24266412 Ha -251.50568722 eV
| XC potential correction : 11.88938413 Ha 323.52660309 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89728931 Ha -51.62786891 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327824 Ha -2078.49475364 eV
| Total energy, T -> 0 : -76.38327824 Ha -2078.49475364 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327824 Ha -2078.49475364 eV
Derived energy quantities:
| Kinetic energy : 76.12213833 Ha 2071.38877517 eV
| Electrostatic energy : -143.26275246 Ha -3898.37784159 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002622 Ha -0.00071346 eV
| Sum of eigenvalues per atom : -376.03021158 eV
| Total energy (T->0) per atom : -692.83158455 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158455 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.828 s, elapsed 3.849 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001987E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.1015E-02
| Change of sum of eigenvalues : 0.1706E+00 eV
| Change of total energy : -0.5262E-05 eV
------------------------------------------------------------
End self-consistency iteration # 6 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.096 s 1.095 s
| Charge density update : 0.488 s 0.489 s
| Density mixing : 0.104 s 0.104 s
| Hartree multipole update : 0.020 s 0.019 s
| Hartree multipole summation : 0.124 s 0.121 s
| Integration : 0.360 s 0.359 s
| Solution of K.-S. eqns. : 0.004 s 0.002 s
| Total energy evaluation : 0.004 s 0.000 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 7
Date : 20210920, Time : 144904.931
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000069
| Charge integration error : 0.0000000069
| Normalization factor for density and gradient : 0.9999999993
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.275421E-12
| Sum of charges compensated after spline to logarithmic grids = 0.131566E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.276011E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.914 s, elapsed 0.932 s
| RMS charge density error from multipole expansion : 0.145904E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.859 s, elapsed 2.870 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.25217406 eV
Highest occupied state (VBM) at -6.97885615 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16286666 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81598950 eV.
Total energy components:
| Sum of eigenvalues : -41.45779781 Ha -1128.12407680 eV
| XC energy correction : -9.24247344 Ha -251.50049845 eV
| XC potential correction : 11.88913443 Ha 323.51980843 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89600105 Ha -51.59281346 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327797 Ha -2078.49474614 eV
| Total energy, T -> 0 : -76.38327797 Ha -2078.49474614 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327797 Ha -2078.49474614 eV
Derived energy quantities:
| Kinetic energy : 76.12260624 Ha 2071.40150765 eV
| Electrostatic energy : -143.26341077 Ha -3898.39575534 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002679 Ha -0.00072895 eV
| Sum of eigenvalues per atom : -376.04135893 eV
| Total energy (T->0) per atom : -692.83158205 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158205 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.829 s, elapsed 3.852 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001915E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.5732E-03
| Change of sum of eigenvalues : -0.3344E-01 eV
| Change of total energy : 0.7497E-05 eV
------------------------------------------------------------
End self-consistency iteration # 7 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.112 s 1.109 s
| Charge density update : 0.492 s 0.489 s
| Density mixing : 0.120 s 0.118 s
| Hartree multipole update : 0.020 s 0.020 s
| Hartree multipole summation : 0.124 s 0.120 s
| Integration : 0.360 s 0.360 s
| Solution of K.-S. eqns. : 0.004 s 0.001 s
| Total energy evaluation : 0.004 s 0.001 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 8
Date : 20210920, Time : 144906.041
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000081
| Charge integration error : 0.0000000081
| Normalization factor for density and gradient : 0.9999999992
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.164239E-12
| Sum of charges compensated after spline to logarithmic grids = 0.131540E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.164313E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.916 s, elapsed 0.934 s
| RMS charge density error from multipole expansion : 0.145847E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.860 s, elapsed 2.870 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.001 s
Finished solving standard eigenproblem
| Time : 0.000 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.25643152 eV
Highest occupied state (VBM) at -6.98209767 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16392219 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81817548 eV.
Total energy components:
| Sum of eigenvalues : -41.45901284 Ha -1128.15713928 eV
| XC energy correction : -9.24229224 Ha -251.49556768 eV
| XC potential correction : 11.88889759 Ha 323.51336351 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89473035 Ha -51.55823599 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327794 Ha -2078.49474530 eV
| Total energy, T -> 0 : -76.38327794 Ha -2078.49474530 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327794 Ha -2078.49474530 eV
Derived energy quantities:
| Kinetic energy : 76.12212615 Ha 2071.38844366 eV
| Electrostatic energy : -143.26311185 Ha -3898.38762128 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002700 Ha -0.00073466 eV
| Sum of eigenvalues per atom : -376.05237976 eV
| Total energy (T->0) per atom : -692.83158177 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158177 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.828 s, elapsed 3.847 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001875E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.1976E-03
| Change of sum of eigenvalues : -0.3306E-01 eV
| Change of total energy : 0.8427E-06 eV
------------------------------------------------------------
End self-consistency iteration # 8 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.124 s 1.123 s
| Charge density update : 0.488 s 0.489 s
| Density mixing : 0.136 s 0.133 s
| Hartree multipole update : 0.020 s 0.019 s
| Hartree multipole summation : 0.124 s 0.121 s
| Integration : 0.360 s 0.359 s
| Solution of K.-S. eqns. : 0.004 s 0.002 s
| Total energy evaluation : 0.000 s 0.000 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 9
Date : 20210920, Time : 144907.165
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000033
| Charge integration error : 0.0000000033
| Normalization factor for density and gradient : 0.9999999997
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.178186E-12
| Sum of charges compensated after spline to logarithmic grids = 0.131527E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.176932E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.920 s, elapsed 0.935 s
| RMS charge density error from multipole expansion : 0.145831E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.936 s, elapsed 2.945 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.25583528 eV
Highest occupied state (VBM) at -6.98208979 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16400835 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81808144 eV.
Total energy components:
| Sum of eigenvalues : -41.45896478 Ha -1128.15583153 eV
| XC energy correction : -9.24229271 Ha -251.49558056 eV
| XC potential correction : 11.88889840 Ha 323.51338576 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89477875 Ha -51.55955310 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327794 Ha -2078.49474529 eV
| Total energy, T -> 0 : -76.38327794 Ha -2078.49474529 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327794 Ha -2078.49474529 eV
Derived energy quantities:
| Kinetic energy : 76.12223228 Ha 2071.39133167 eV
| Electrostatic energy : -143.26321751 Ha -3898.39049640 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002701 Ha -0.00073504 eV
| Sum of eigenvalues per atom : -376.05194384 eV
| Total energy (T->0) per atom : -692.83158176 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158176 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.829 s, elapsed 3.851 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001877E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.3973E-04
| Change of sum of eigenvalues : 0.1308E-02 eV
| Change of total energy : 0.1353E-07 eV
------------------------------------------------------------
End self-consistency iteration # 9 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.148 s 1.147 s
| Charge density update : 0.488 s 0.489 s
| Density mixing : 0.148 s 0.147 s
| Hartree multipole update : 0.020 s 0.019 s
| Hartree multipole summation : 0.124 s 0.121 s
| Integration : 0.372 s 0.369 s
| Solution of K.-S. eqns. : 0.004 s 0.001 s
| Total energy evaluation : 0.004 s 0.000 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 10
Date : 20210920, Time : 144908.313
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000087
| Charge integration error : 0.0000000087
| Normalization factor for density and gradient : 0.9999999991
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.283956E-12
| Sum of charges compensated after spline to logarithmic grids = 0.131527E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.284596E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.909 s, elapsed 0.926 s
| RMS charge density error from multipole expansion : 0.145831E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.936 s, elapsed 2.944 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.25591189 eV
Highest occupied state (VBM) at -6.98211179 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16401751 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81809428 eV.
Total energy components:
| Sum of eigenvalues : -41.45897840 Ha -1128.15620213 eV
| XC energy correction : -9.24229063 Ha -251.49552394 eV
| XC potential correction : 11.88889567 Ha 323.51331129 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89476447 Ha -51.55916464 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327794 Ha -2078.49474529 eV
| Total energy, T -> 0 : -76.38327794 Ha -2078.49474529 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327794 Ha -2078.49474529 eV
Derived energy quantities:
| Kinetic energy : 76.12221606 Ha 2071.39089013 eV
| Electrostatic energy : -143.26320337 Ha -3898.39011148 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002702 Ha -0.00073517 eV
| Sum of eigenvalues per atom : -376.05206738 eV
| Total energy (T->0) per atom : -692.83158176 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158176 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.827 s, elapsed 3.850 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001877E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.3647E-05
| Change of sum of eigenvalues : -0.3706E-03 eV
| Change of total energy : 0.2320E-11 eV
------------------------------------------------------------
End self-consistency iteration # 10 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.164 s 1.160 s
| Charge density update : 0.492 s 0.489 s
| Density mixing : 0.164 s 0.161 s
| Hartree multipole update : 0.020 s 0.020 s
| Hartree multipole summation : 0.120 s 0.120 s
| Integration : 0.368 s 0.368 s
| Solution of K.-S. eqns. : 0.004 s 0.002 s
| Total energy evaluation : 0.000 s 0.000 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 11
Date : 20210920, Time : 144909.474
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000041
| Charge integration error : 0.0000000041
| Normalization factor for density and gradient : 0.9999999996
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.143921E-12
| Sum of charges compensated after spline to logarithmic grids = 0.131527E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.144979E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.916 s, elapsed 0.934 s
| RMS charge density error from multipole expansion : 0.145831E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.937 s, elapsed 2.945 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.25590819 eV
Highest occupied state (VBM) at -6.98210999 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16401841 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81809158 eV.
Total energy components:
| Sum of eigenvalues : -41.45897785 Ha -1128.15618715 eV
| XC energy correction : -9.24229068 Ha -251.49552537 eV
| XC potential correction : 11.88889573 Ha 323.51331308 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89476504 Ha -51.55917997 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327794 Ha -2078.49474529 eV
| Total energy, T -> 0 : -76.38327794 Ha -2078.49474529 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327794 Ha -2078.49474529 eV
Derived energy quantities:
| Kinetic energy : 76.12221700 Ha 2071.39091587 eV
| Electrostatic energy : -143.26320426 Ha -3898.39013578 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002702 Ha -0.00073518 eV
| Sum of eigenvalues per atom : -376.05206238 eV
| Total energy (T->0) per atom : -692.83158176 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158176 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 3.826 s, elapsed 3.848 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001876E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.4999E-06
| Change of sum of eigenvalues : 0.1497E-04 eV
| Change of total energy : -0.2011E-10 eV
------------------------------------------------------------
End self-consistency iteration # 11 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.172 s 1.168 s
| Charge density update : 0.488 s 0.488 s
| Density mixing : 0.172 s 0.169 s
| Hartree multipole update : 0.020 s 0.019 s
| Hartree multipole summation : 0.124 s 0.121 s
| Integration : 0.372 s 0.369 s
| Solution of K.-S. eqns. : 0.004 s 0.001 s
| Total energy evaluation : 0.004 s 0.001 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 12
Date : 20210920, Time : 144910.643
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000077
| Charge integration error : 0.0000000077
| Normalization factor for density and gradient : 0.9999999992
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.108624E-12
| Sum of charges compensated after spline to logarithmic grids = 0.131527E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.108771E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.913 s, elapsed 0.931 s
| RMS charge density error from multipole expansion : 0.145831E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 2.936 s, elapsed 2.945 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.25590966 eV
Highest occupied state (VBM) at -6.98210776 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16401632 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81809144 eV.
Total energy components:
| Sum of eigenvalues : -41.45897711 Ha -1128.15616716 eV
| XC energy correction : -9.24229088 Ha -251.49553066 eV
| XC potential correction : 11.88889598 Ha 323.51331985 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89476583 Ha -51.55920145 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327794 Ha -2078.49474529 eV
| Total energy, T -> 0 : -76.38327794 Ha -2078.49474529 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327794 Ha -2078.49474529 eV
Derived energy quantities:
| Kinetic energy : 76.12221669 Ha 2071.39090724 eV
| Electrostatic energy : -143.26320375 Ha -3898.39012186 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002702 Ha -0.00073518 eV
| Sum of eigenvalues per atom : -376.05205572 eV
| Total energy (T->0) per atom : -692.83158176 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158176 eV
Evaluating new KS density using the density matrix
Evaluating density matrix
Time summed over all CPUs for getting density from density matrix: real work 4.483 s, elapsed 4.504 s
Integration grid: deviation in total charge (<rho> - N_e) = 1.001876E-08
Self-consistency convergence accuracy:
| Change of charge density : 0.4059E-06
| Change of sum of eigenvalues : 0.1999E-04 eV
| Change of total energy : -0.8353E-10 eV
------------------------------------------------------------
End self-consistency iteration # 12 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 1.252 s 1.250 s
| Charge density update : 0.572 s 0.570 s
| Density mixing : 0.172 s 0.169 s
| Hartree multipole update : 0.020 s 0.019 s
| Hartree multipole summation : 0.124 s 0.120 s
| Integration : 0.372 s 0.368 s
| Solution of K.-S. eqns. : 0.004 s 0.002 s
| Total energy evaluation : 0.000 s 0.001 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
------------------------------------------------------------
Begin self-consistency iteration # 13
Date : 20210920, Time : 144911.894
------------------------------------------------------------
Pulay mixing of updated and previous charge densities.
Renormalizing the density to the exact electron count on the 3D integration grid.
| Formal number of electrons (from input files) : 10.0000000000
| Integrated number of electrons on 3D grid : 10.0000000098
| Charge integration error : 0.0000000098
| Normalization factor for density and gradient : 0.9999999990
Evaluating partitioned Hartree potential by multipole expansion.
| Original multipole sum: apparent total charge = 0.179121E-12
| Sum of charges compensated after spline to logarithmic grids = 0.131527E-06
| Analytical far-field extrapolation by fixed multipoles:
| Hartree multipole sum: apparent total charge = 0.179268E-12
Summing up the Hartree potential.
Time summed over all CPUs for potential: real work 0.959 s, elapsed 0.979 s
| RMS charge density error from multipole expansion : 0.145831E-02
Integrating Hamiltonian matrix: batch-based integration.
Time summed over all CPUs for integration: real work 3.399 s, elapsed 3.410 s
Updating Kohn-Sham eigenvalues and eigenvectors using ELSI and the (modified) LAPACK eigensolver.
Starting LAPACK eigensolver
Finished Cholesky decomposition
| Time : 0.000 s
Finished transformation to standard eigenproblem
| Time : 0.000 s
Finished solving standard eigenproblem
| Time : 0.001 s
Finished back-transformation of eigenvectors
| Time : 0.000 s
Obtaining occupation numbers and chemical potential using ELSI.
| Chemical potential (Fermi level): -3.25590890 eV
Highest occupied state (VBM) at -6.98210735 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16401618 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81809117 eV.
Total energy components:
| Sum of eigenvalues : -41.45897693 Ha -1128.15616212 eV
| XC energy correction : -9.24229090 Ha -251.49553132 eV
| XC potential correction : 11.88889601 Ha 323.51332072 eV
| Free-atom electrostatic energy: -35.67614010 Ha -970.79716586 eV
| Hartree energy correction : -1.89476602 Ha -51.55920670 eV
| Entropy correction : 0.00000000 Ha 0.00000000 eV
| ---------------------------
| Total energy : -76.38327794 Ha -2078.49474529 eV
| Total energy, T -> 0 : -76.38327794 Ha -2078.49474529 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -76.38327794 Ha -2078.49474529 eV
Derived energy quantities:
| Kinetic energy : 76.12221677 Ha 2071.39090963 eV
| Electrostatic energy : -143.26320381 Ha -3898.39012359 eV
| Energy correction for multipole
| error in Hartree potential : -0.00002702 Ha -0.00073518 eV
| Sum of eigenvalues per atom : -376.05205404 eV
| Total energy (T->0) per atom : -692.83158176 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy per atom : -692.83158176 eV
Self-consistency convergence accuracy:
| Change of charge density : 0.3728E-07
| Change of sum of eigenvalues : 0.5044E-05 eV
| Change of total energy : -0.2320E-11 eV
Writing Kohn-Sham eigenvalues.
State Occupation Eigenvalue [Ha] Eigenvalue [eV]
1 2.00000 -18.744255 -510.05713
2 2.00000 -0.920017 -25.03494
3 2.00000 -0.473935 -12.89643
4 2.00000 -0.334694 -9.10748
5 2.00000 -0.256588 -6.98211
6 0.00000 -0.006027 -0.16402
7 0.00000 0.063928 1.73956
8 0.00000 0.269211 7.32559
9 0.00000 0.301997 8.21775
10 0.00000 0.311873 8.48650
11 0.00000 0.388652 10.57575
12 0.00000 0.432486 11.76854
13 0.00000 0.437829 11.91395
14 0.00000 0.477880 13.00379
15 0.00000 0.541407 14.73244
16 0.00000 0.642811 17.49178
17 0.00000 0.745005 20.27263
18 0.00000 0.889092 24.19344
19 0.00000 0.924311 25.15178
20 0.00000 1.132966 30.82958
21 0.00000 1.169546 31.82496
22 0.00000 1.219484 33.18386
23 0.00000 1.223580 33.29530
24 0.00000 1.315779 35.80417
25 0.00000 1.376231 37.44916
26 0.00000 1.591369 43.30336
27 0.00000 1.789932 48.70653
28 0.00000 1.905835 51.86042
29 0.00000 1.995271 54.29409
30 0.00000 2.066031 56.21957
31 0.00000 2.100776 57.16502
32 0.00000 2.213085 60.22112
33 0.00000 2.215269 60.28053
34 0.00000 2.242405 61.01894
35 0.00000 2.306540 62.76414
36 0.00000 2.348518 63.90644
37 0.00000 2.380978 64.78970
38 0.00000 2.493668 67.85615
39 0.00000 2.541009 69.14438
40 0.00000 2.674868 72.78685
41 0.00000 2.792145 75.97813
42 0.00000 2.941975 80.05522
43 0.00000 2.968299 80.77152
44 0.00000 3.062599 83.33757
45 0.00000 3.227885 87.83523
46 0.00000 3.397295 92.44510
47 0.00000 3.558800 96.83987
48 0.00000 3.671503 99.90667
49 0.00000 3.743973 101.87868
50 0.00000 3.769948 102.58550
51 0.00000 4.000471 108.85836
52 0.00000 4.022622 109.46112
53 0.00000 4.210366 114.56990
54 0.00000 4.279292 116.44547
55 0.00000 4.365494 118.79114
56 0.00000 4.449803 121.08530
57 0.00000 4.853656 132.07471
58 0.00000 4.921291 133.91515
59 0.00000 5.591996 152.16595
60 0.00000 5.692986 154.91404
61 0.00000 5.853123 159.27158
62 0.00000 5.921379 161.12892
63 0.00000 6.255053 170.20866
64 0.00000 6.345838 172.67904
65 0.00000 6.589603 179.31222
66 0.00000 6.769485 184.20706
67 0.00000 6.849210 186.37649
68 0.00000 6.920337 188.31195
69 0.00000 6.979055 189.90976
70 0.00000 7.038339 191.52296
71 0.00000 7.111550 193.51512
72 0.00000 7.145886 194.44945
73 0.00000 7.556626 205.62625
74 0.00000 7.562912 205.79731
75 0.00000 7.633631 207.72166
76 0.00000 7.662936 208.51911
77 0.00000 7.708480 209.75842
78 0.00000 7.809368 212.50370
79 0.00000 7.819561 212.78109
80 0.00000 7.888314 214.65196
81 0.00000 7.939890 216.05540
82 0.00000 8.148314 221.72690
83 0.00000 8.289784 225.57650
84 0.00000 8.377751 227.97020
85 0.00000 8.383835 228.13576
86 0.00000 8.670348 235.93217
87 0.00000 8.777955 238.86032
88 0.00000 8.896164 242.07693
89 0.00000 9.285395 252.66847
90 0.00000 9.393599 255.61284
91 0.00000 9.629707 262.03766
92 0.00000 9.800501 266.68520
93 0.00000 9.820914 267.24067
94 0.00000 10.089010 274.53593
95 0.00000 10.133849 275.75607
96 0.00000 10.598991 288.41321
97 0.00000 10.726556 291.88443
98 0.00000 10.840395 294.98215
99 0.00000 10.946592 297.87193
100 0.00000 10.955152 298.10484
101 0.00000 11.124680 302.71796
102 0.00000 11.434477 311.14796
103 0.00000 11.535390 313.89392
104 0.00000 11.657905 317.22772
105 0.00000 11.658510 317.24419
106 0.00000 11.752103 319.79099
107 0.00000 12.817891 348.79257
108 0.00000 13.042406 354.90193
109 0.00000 13.476860 366.72401
110 0.00000 13.823335 376.15207
111 0.00000 13.857224 377.07425
112 0.00000 13.967211 380.06716
113 0.00000 15.615551 424.92077
114 0.00000 15.970352 434.57539
115 0.00000 42.382645 1153.29046
Highest occupied state (VBM) at -6.98210735 eV
| Occupation number: 2.00000000
Lowest unoccupied state (CBM) at -0.16401618 eV
| Occupation number: 0.00000000
Overall HOMO-LUMO gap: 6.81809117 eV.
| Chemical Potential : -3.25590890 eV
Self-consistency cycle converged.
------------------------------------------------------------
End self-consistency iteration # 13 : max(cpu_time) wall_clock(cpu1)
| Time for this iteration : 0.744 s 0.746 s
| Charge density update : 0.000 s 0.000 s
| Density mixing : 0.168 s 0.168 s
| Hartree multipole update : 0.020 s 0.020 s
| Hartree multipole summation : 0.128 s 0.126 s
| Integration : 0.428 s 0.427 s
| Solution of K.-S. eqns. : 0.004 s 0.002 s
| Total energy evaluation : 0.004 s 0.000 s
Partial memory accounting:
| Current value for overall tracked memory usage:
| Minimum: 0.205 MB (on task 0)
| Maximum: 0.205 MB (on task 0)
| Average: 0.205 MB
| Peak value for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation so far:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
------------------------------------------------------------
Energy and forces in a compact form:
| Total energy uncorrected : -0.207849474528721E+04 eV
| Total energy corrected : -0.207849474528721E+04 eV <-- do not rely on this value for anything but (periodic) metals
| Electronic free energy : -0.207849474528721E+04 eV
------------------------------------
Start decomposition of the XC Energy
------------------------------------
X and C from original XC functional choice
Hartree-Fock Energy : 0.000000000 Ha 0.000000000 eV
X Energy : -8.917081740 Ha -242.646139838 eV
C Energy : -0.325209160 Ha -8.849391487 eV
Total XC Energy : -9.242290899 Ha -251.495531325 eV
------------------------------------
LDA X and C from self-consistent density
X Energy LDA : -8.102291018 Ha -220.474556210 eV
C Energy LDA : -0.659919566 Ha -17.957325053 eV
------------------------------------
End decomposition of the XC Energy
------------------------------------
------------------------------------------------------------
--------------------------------------------
Constructing auxiliary basis (full product) ...
Product basis:
| charge radius: extent of product basis function
| field radius: extent of its Coulomb potential
| Species l charge radius field radius multipol momen
| H 0 1.540738 A 1.540738 A -6.916037E-18 a.u.
| H 1 1.719935 A 1.719935 A -4.755617E-17 a.u.
| H 2 1.741090 A 1.741090 A 1.289987E-17 a.u.
| H 3 1.850834 A 1.850834 A -2.647845E-17 a.u.
| H 1 1.943589 A 1.943589 A 2.065226E-17 a.u.
| H 4 1.943589 A 1.943589 A -5.492333E-17 a.u.
| H 3 2.091510 A 2.091510 A 6.297030E-17 a.u.
| H 5 2.250689 A 2.250689 A -5.747961E-17 a.u.
| H 0 2.306396 A 2.306396 A 2.774351E-17 a.u.
| H 2 2.392554 A 2.392554 A -7.469263E-17 a.u.
| H 1 2.421982 A 2.421982 A -2.277055E-16 a.u.
| H 3 2.606312 A 2.606312 A 3.242236E-16 a.u.
| H 4 2.736928 A 2.736928 A 8.717519E-17 a.u.
| H 4 2.804670 A 2.804670 A 3.481406E-16 a.u.
| H 0 2.874090 A 2.874090 A -2.361050E-16 a.u.
| H 2 2.909441 A 2.909441 A -1.809227E-16 a.u.
| H 1 2.945227 A 2.945227 A -8.821544E-18 a.u.
| H 3 3.247826 A 3.247826 A -2.697485E-16 a.u.
| H 2 3.998065 A 3.998065 A -1.652068E-16 a.u.
| H 2 3.998065 A 3.998065 A 4.436768E-16 a.u.
| H 1 4.047241 A 4.047241 A 5.675197E-16 a.u.
| H 0 4.097023 A 4.097023 A -2.604829E-16 a.u.
| H 1 4.921608 A 4.921608 A 7.071742E-16 a.u.
| H 0 5.043424 A 5.043424 A -2.528286E-16 a.u.
| H 0 6.846310 A 6.846310 A 2.565054E-16 a.u.
| H 5 2.306396 A 9.019552E+01 A 1.951161 a.u.
| H 4 2.839168 A 2.911229E+02 A 4.010204 a.u.
| H 3 3.287774 A 1.327409E+03 A 3.150680 a.u.
| H 2 4.047241 A 1.550050E+04 A 1.999973 a.u.
| H 1 4.921608 A 2.036188E+06 A 1.178213 a.u.
| H 0 6.846310 A 4.106636E+12 A 0.617554 a.u.
| O 0 0.933362 A 0.933362 A -7.103052E-17 a.u.
| O 0 0.956464 A 0.956464 A -2.967789E-17 a.u.
| O 1 0.992193 A 0.992193 A -1.156826E-17 a.u.
| O 0 1.206546 A 1.206546 A -2.371352E-16 a.u.
| O 1 1.251618 A 1.251618 A 2.323404E-17 a.u.
| O 2 1.267013 A 1.267013 A 2.664443E-18 a.u.
| O 3 1.363441 A 1.363441 A 1.396047E-17 a.u.
| O 1 1.397188 A 1.397188 A 1.586828E-17 a.u.
| O 4 1.431771 A 1.431771 A 6.233050E-18 a.u.
| O 3 1.559690 A 1.559690 A 1.717151E-17 a.u.
| O 5 1.637854 A 1.637854 A -7.268710E-17 a.u.
| O 0 1.741090 A 1.741090 A 3.949007E-17 a.u.
| O 2 1.762506 A 1.762506 A 2.581467E-17 a.u.
| O 1 1.806130 A 1.806130 A 1.712499E-17 a.u.
| O 5 1.873600 A 1.873600 A 1.484932E-16 a.u.
| O 3 1.919974 A 1.919974 A -1.026672E-16 a.u.
| O 4 1.967496 A 1.967496 A -6.265784E-17 a.u.
| O 0 2.143278 A 2.143278 A 4.311560E-17 a.u.
| O 1 2.169640 A 2.169640 A 5.779930E-17 a.u.
| O 2 2.169640 A 2.169640 A 5.336136E-17 a.u.
| O 5 2.250689 A 2.250689 A -8.173302E-16 a.u.
| O 3 2.363483 A 2.363483 A -1.863301E-16 a.u.
| O 4 2.451772 A 2.451772 A -1.795346E-16 a.u.
| O 3 2.670821 A 2.670821 A 6.421252E-16 a.u.
| O 0 2.839168 A 2.839168 A -1.196742E-16 a.u.
| O 5 2.874090 A 2.874090 A -1.549761E-15 a.u.
| O 1 2.945227 A 2.945227 A -2.505802E-16 a.u.
| O 2 2.981454 A 2.981454 A 7.352719E-17 a.u.
| O 3 3.247826 A 3.247826 A 1.892654E-15 a.u.
| O 4 3.369151 A 3.369151 A -7.056860E-16 a.u.
| O 4 3.369151 A 3.369151 A 2.101640E-15 a.u.
| O 0 3.452542 A 3.452542 A 6.658395E-17 a.u.
| O 1 3.581514 A 3.581514 A -1.940867E-16 a.u.
| O 2 3.670161 A 3.670161 A 1.040913E-15 a.u.
| O 3 4.047241 A 4.047241 A -4.244186E-16 a.u.
| O 0 4.629785 A 4.629785 A 3.592314E-16 a.u.
| O 0 4.686731 A 4.686731 A 4.842889E-16 a.u.
| O 1 4.861808 A 4.861808 A 3.549878E-16 a.u.
| O 2 5.043424 A 5.043424 A -5.730093E-16 a.u.
| O 2 5.105458 A 5.105458 A -1.710411E-15 a.u.
| O 2 5.231825 A 5.231825 A 6.715257E-16 a.u.
| O 5 2.909441 A 1.208207E+02 A 11.272765 a.u.
| O 4 3.452542 A 3.360361E+02 A 8.217017 a.u.
| O 3 4.097023 A 1.653479E+03 A 7.585427 a.u.
| O 2 5.231825 A 1.996977E+04 A 4.276672 a.u.
| O 1 4.861808 A 1.981796E+06 A 1.116107 a.u.
| O 0 4.629785 A 3.514475E+12 A 0.528505 a.u.
| Shrink_full_auxil_basis : there are 78 radial auxiliary wave functions
accepted and 305 rejected.
| Shrink_full_auxil_basis : there are totally 549 partial auxiliary wave functions.
| Number of product basis functions per thread : 69
| Minimal requirement for computer memory : 0.030 Gbs
Basis pair condensation : 6670 --> 6670
----------------------------------------------------
Integrating the 3-basis-function Coulomb matrix ...
| i_atom: 1
| i_atom: 2
| i_atom: 3
Integrating the Coulomb interaction matrix for auxiliary basis functions (by atoms)
SBT integration errors (all should be 'small'):
large logFT aliasing -> increase N/lnrange
large SBT aliasing -> decrease lnk0 & increase lnk0+lnrange
large SBT ringing -> decrease lnr0 & increase lnr0+lnrange
| El't L: logFT-al. small-k large-k small-r large-r
Atomic logSBT for 2-center Coulomb matrix : max(cpu_time) wall_clock(cpu1)
| Multiplication with kernel : 0.008 s 0.005 s
| Main matrix multiplication : 0.028 s 0.014 s
| Analytic angular integration : 0.008 s 0.002 s
| Kernel construction : 0.008 s 0.003 s
| Overall 2-center overlap time : 0.032 s 0.025 s
Difference of Coulomb matrix to its transposed: 3.0531E-16
Task 0: Eigenvalues of the Coulomb matrix range from 2.9999E+00 to 1.2448E-07.
Task 0: Using 539 eigenvalues out of rank 549 Coulomb matrix (auxiliary basis).
Task 0: Still using eigenvalue 8.9494E-05 while cutting 6.5911E-06 in Coulomb matrix.
Multiplying V^-0.5 x ovlp_3fn (1d-scalapack)
End of correlation preparation : max(cpu_time) wall_clock(cpu1)
| Product basis setup: Total time : 42.528 s 42.526 s
| Product basis: | 3-center integrations : 42.080 s 42.088 s
| Product basis: | 2-center integrations : 0.088 s 0.085 s
| Product basis: | 2-center linear algebra : 0.152 s 0.149 s
| Product basis: | 3-center x 2-center : 0.032 s 0.030 s
--------------------------------------------
GW quasiparticle calculation starts ...
The ovlp_3KS matrix takes another 1 MiB x 8 procs = 0.007 GiB.
Initialising transformed Gauss-Legendre time and frequency grids
Number of frequency points for self energy.....:200
Number of frequency points....................:200
Starts to calculate the exchange energy ...
Integrating the xc potential matrix for basis functions ...
-------------------------------------------------
Start to calculate the self energy ...
HOMO and first non-fully-occupied orbitals: 5 6
| i_freq 1 0.000018
| i_freq 2 0.000095
| i_freq 3 0.000233
| i_freq 4 0.000433
| i_freq 5 0.000694
| i_freq 6 0.001017
| i_freq 7 0.001402
| i_freq 8 0.001849
| i_freq 9 0.002358
| i_freq 10 0.002930
| i_freq 11 0.003564
| i_freq 12 0.004262
| i_freq 13 0.005023
| i_freq 14 0.005848
| i_freq 15 0.006737
| i_freq 16 0.007691
| i_freq 17 0.008711
| i_freq 18 0.009796
| i_freq 19 0.010947
| i_freq 20 0.012165
| i_freq 21 0.013451
| i_freq 22 0.014804
| i_freq 23 0.016227
| i_freq 24 0.017719
| i_freq 25 0.019281
| i_freq 26 0.020915
| i_freq 27 0.022620
| i_freq 28 0.024398
| i_freq 29 0.026250
| i_freq 30 0.028177
| i_freq 31 0.030180
| i_freq 32 0.032259
| i_freq 33 0.034417
| i_freq 34 0.036654
| i_freq 35 0.038971
| i_freq 36 0.041369
| i_freq 37 0.043851
| i_freq 38 0.046417
| i_freq 39 0.049069
| i_freq 40 0.051808
| i_freq 41 0.054635
| i_freq 42 0.057554
| i_freq 43 0.060564
| i_freq 44 0.063668
| i_freq 45 0.066868
| i_freq 46 0.070165
| i_freq 47 0.073562
| i_freq 48 0.077060
| i_freq 49 0.080662
| i_freq 50 0.084369
| i_freq 51 0.088185
| i_freq 52 0.092111
| i_freq 53 0.096150
| i_freq 54 0.100304
| i_freq 55 0.104576
| i_freq 56 0.108969
| i_freq 57 0.113486
| i_freq 58 0.118130
| i_freq 59 0.122903
| i_freq 60 0.127809
| i_freq 61 0.132851
| i_freq 62 0.138032
| i_freq 63 0.143357
| i_freq 64 0.148829
| i_freq 65 0.154452
| i_freq 66 0.160229
| i_freq 67 0.166165
| i_freq 68 0.172265
| i_freq 69 0.178533
| i_freq 70 0.184973
| i_freq 71 0.191590
| i_freq 72 0.198390
| i_freq 73 0.205378
| i_freq 74 0.212559
| i_freq 75 0.219939
| i_freq 76 0.227524
| i_freq 77 0.235321
| i_freq 78 0.243335
| i_freq 79 0.251573
| i_freq 80 0.260044
| i_freq 81 0.268753
| i_freq 82 0.277708
| i_freq 83 0.286918
| i_freq 84 0.296392
| i_freq 85 0.306136
| i_freq 86 0.316162
| i_freq 87 0.326478
| i_freq 88 0.337094
| i_freq 89 0.348020
| i_freq 90 0.359268
| i_freq 91 0.370850
| i_freq 92 0.382775
| i_freq 93 0.395059
| i_freq 94 0.407712
| i_freq 95 0.420750
| i_freq 96 0.434186
| i_freq 97 0.448035
| i_freq 98 0.462314
| i_freq 99 0.477039
| i_freq 100 0.492227
| i_freq 101 0.507896
| i_freq 102 0.524067
| i_freq 103 0.540758
| i_freq 104 0.557992
| i_freq 105 0.575790
| i_freq 106 0.594177
| i_freq 107 0.613178
| i_freq 108 0.632817
| i_freq 109 0.653124
| i_freq 110 0.674128
| i_freq 111 0.695858
| i_freq 112 0.718349
| i_freq 113 0.741634
| i_freq 114 0.765749
| i_freq 115 0.790734
| i_freq 116 0.816630
| i_freq 117 0.843479
| i_freq 118 0.871328
| i_freq 119 0.900225
| i_freq 120 0.930224
| i_freq 121 0.961378
| i_freq 122 0.993746
| i_freq 123 1.027391
| i_freq 124 1.062380
| i_freq 125 1.098784
| i_freq 126 1.136679
| i_freq 127 1.176144
| i_freq 128 1.217269
| i_freq 129 1.260144
| i_freq 130 1.304869
| i_freq 131 1.351551
| i_freq 132 1.400305
| i_freq 133 1.451252
| i_freq 134 1.504525
| i_freq 135 1.560267
| i_freq 136 1.618630
| i_freq 137 1.679780
| i_freq 138 1.743896
| i_freq 139 1.811170
| i_freq 140 1.881813
| i_freq 141 1.956051
| i_freq 142 2.034131
| i_freq 143 2.116320
| i_freq 144 2.202911
| i_freq 145 2.294221
| i_freq 146 2.390599
| i_freq 147 2.492424
| i_freq 148 2.600113
| i_freq 149 2.714123
| i_freq 150 2.834958
| i_freq 151 2.963169
| i_freq 152 3.099370
| i_freq 153 3.244234
| i_freq 154 3.398512
| i_freq 155 3.563035
| i_freq 156 3.738727
| i_freq 157 3.926622
| i_freq 158 4.127874
| i_freq 159 4.343777
| i_freq 160 4.575787
| i_freq 161 4.825544
| i_freq 162 5.094902
| i_freq 163 5.385966
| i_freq 164 5.701130
| i_freq 165 6.043126
| i_freq 166 6.415087
| i_freq 167 6.820615
| i_freq 168 7.263872
| i_freq 169 7.749683
| i_freq 170 8.283674
| i_freq 171 8.872428
| i_freq 172 9.523696
| i_freq 173 10.246646
| i_freq 174 11.052196
| i_freq 175 11.953420
| i_freq 176 12.966087
| i_freq 177 14.109356
| i_freq 178 15.406685
| i_freq 179 16.887044
| i_freq 180 18.586551
| i_freq 181 20.550690
| i_freq 182 22.837391
| i_freq 183 25.521346
| i_freq 184 28.700187
| i_freq 185 32.503494
| i_freq 186 37.106206
| i_freq 187 42.749103
| i_freq 188 49.770942
| i_freq 189 58.660500
| i_freq 190 70.143974
| i_freq 191 85.338099
| i_freq 192 106.032192
| i_freq 193 135.239901
| i_freq 194 178.361304
| i_freq 195 245.869090
| i_freq 196 360.317302
| i_freq 197 577.921171
| i_freq 198 1073.293071
| i_freq 199 2638.240217
| i_freq 200 13902.153495
----------------------------------------------------------------------------------------
Analytical continuation starts... [n_pade_params = 16]
Quasi particle energy calculation using analytic continuation starts...
----------------------------------------------------------------------------------------
GW quasi-particle energy levels
e_qp = e_gs + e_x^ex - e_xc^gs + e_c^nloc
state occ_num e_gs e_x^ex e_xc^gs e_c^nloc e_qp
----------------------------------------------------------------------------------------
----------------------------------------------------------------------------------------
1 2.0000 -510.0571 -132.6661 -83.7896 10.2278 -548.7059
2 2.0000 -25.0349 -33.0815 -21.4599 5.9990 -30.6576
3 2.0000 -12.8964 -24.7466 -18.2592 0.9986 -18.3851
4 2.0000 -9.1075 -25.8610 -18.9948 1.7442 -14.2295
5 2.0000 -6.9821 -26.1996 -19.2531 1.8777 -12.0509
6 0.0000 -0.1640 -2.7787 -6.4712 -0.8678 2.6608
7 0.0000 1.7396 -2.7882 -6.2767 -0.8666 4.3615
8 0.0000 7.3256 -3.1214 -7.8597 -1.2802 10.7837
9 0.0000 8.2178 -3.2342 -8.2847 -1.5524 11.7158
10 0.0000 8.4865 -3.1617 -7.6384 -1.2360 11.7271
11 0.0000 10.5757 -4.5567 -9.9114 -1.4943 14.4362
12 0.0000 11.7685 -1.9066 -6.4850 -1.2193 15.1276
13 0.0000 11.9139 -2.7382 -7.4045 -1.6776 14.9027
14 0.0000 13.0038 -1.8283 -6.6738 -1.2680 16.5813
15 0.0000 14.7324 -1.6680 -6.3771 -1.1389 18.3026
----------------------------------------------------------------------------------------
DFT/Hartree-Fock HOMO level (eV): -6.9821 -6.9821
Quasiparticle HOMO level (eV): -12.0509 -12.0509
----------------------------------------------------------------------------------------
| Total time for transforming the 3-center integrals : 0.008 s
| Total time for calculating the exchange-correlation
energy matrix elements : 0.576 s
| Total time for calculating polarisability
for imaginary frequencies : 1.816 s
| Total time for calculating self energy
on imaginary frequency axis : 37.688 s
| Total time for calculating the quasiparticle
energies with analytic continuation : 0.004 s
| Total time for calculating the quasiparticle
energies with contour deformation : 0.000 s
------------------------------------------------------------------------------
Final output of selected total energy values:
The following output summarizes some interesting total energy values
at the end of a run (AFTER all relaxation, molecular dynamics, etc.).
| Total energy of the DFT / Hartree-Fock s.c.f. calculation : -2078.494745287 eV
| Final zero-broadening corrected energy (caution - metals only) : -2078.494745287 eV
| For reference only, the value of 1 Hartree used in FHI-aims is : 27.211384500 eV
Before relying on these values, please be sure to understand exactly which
total energy value is referred to by a given number. Different objects may
all carry the same name 'total energy'. Definitions:
Total energy of the DFT / Hartree-Fock s.c.f. calculation:
| Note that this energy does not include ANY quantities calculated after the
| s.c.f. cycle, in particular not ANY RPA, MP2, etc. many-body perturbation terms.
Final zero-broadening corrected energy:
| For metallic systems only, a broadening of the occupation numbers at the Fermi
| level can be extrapolated back to zero broadening by an electron-gas inspired
| formula. For all systems that are not real metals, this value can be
| meaningless and should be avoided.
------------------------------------------------------------------------------
Methods described in the following list of references were used in this FHI-aims run.
If you publish the results, please make sure to cite these reference if they apply.
FHI-aims is an academic code, and for our developers (often, Ph.D. students
and postdocs), scientific credit in the community is essential.
Thank you for helping us!
For any use of FHI-aims, please cite:
Volker Blum, Ralf Gehrke, Felix Hanke, Paula Havu, Ville Havu,
Xinguo Ren, Karsten Reuter, and Matthias Scheffler
'Ab initio molecular simulations with numeric atom-centered orbitals'
Computer Physics Communications 180, 2175-2196 (2009)
http://dx.doi.org/10.1016/j.cpc.2009.06.022
For Hartree-Fock, hybrid functionals, or many-body perturbation theory used in your run, please cite:
Xinguo Ren, Patrick Rinke, Volker Blum, Juergen Wieferink, Alex Tkatchenko,
Andrea Sanfilippo, Karsten Reuter, and Matthias Scheffler,
'Resolution-of-identity approach to Hartree-Fock, hybrid density functionals,
RPA, MP2, and GW with numeric atom-centered orbital basis functions'
New Journal of Physics 14, 053020 (2012).
http://dx.doi.org/10.1088/1367-2630/14/5/053020
The ELSI infrastructure was used in your run to solve the Kohn-Sham electronic structure.
Please check out http://elsi-interchange.org to learn more.
If scalability is important for your project, please acknowledge ELSI by citing:
V. W-z. Yu, F. Corsetti, A. Garcia, W. P. Huhn, M. Jacquelin, W. Jia,
B. Lange, L. Lin, J. Lu, W. Mi, A. Seifitokaldani, A. Vazquez-Mayagoitia,
C. Yang, H. Yang, and V. Blum
'ELSI: A unified software interface for Kohn-Sham electronic structure solvers'
Computer Physics Communications 222, 267-285 (2018).
http://dx.doi.org/10.1016/j.cpc.2017.09.007
For the real-space grid partitioning and parallelization used in this calculation, please cite:
Ville Havu, Volker Blum, Paula Havu, and Matthias Scheffler,
'Efficient O(N) integration for all-electron electronic structure calculation'
'using numerically tabulated basis functions'
Journal of Computational Physics 228, 8367-8379 (2009).
http://dx.doi.org/10.1016/j.jcp.2009.08.008
Of course, there are many other important community references, e.g., those cited in the
above references. Our list is limited to references that describe implementations in the
FHI-aims code. The reason is purely practical (length of this list) - please credit others as well.
------------------------------------------------------------
Leaving FHI-aims.
Date : 20210920, Time : 145035.399
Computational steps:
| Number of self-consistency cycles : 13
| Number of SCF (re)initializations : 1
Detailed time accounting : max(cpu_time) wall_clock(cpu1)
| Total time : 103.264 s 106.144 s
| Preparation time : 0.372 s 2.049 s
| Boundary condition initalization : 0.000 s 0.001 s
| Grid partitioning : 0.244 s 0.243 s
| Preloading free-atom quantities on grid : 0.220 s 0.276 s
| Free-atom superposition energy : 0.068 s 0.066 s
| Total time for integrations : 9.440 s 9.424 s
| Product basis setup: Total time : 0.000 s 0.000 s
| Product basis: | 3-center integrations :( 42.080 s) ( 42.088 s)
| Product basis: | 2-center integrations :( 0.088 s) ( 0.085 s)
| Product basis: | 2-center linear algebra :( 0.152 s) ( 0.149 s)
| Product basis: | 3-center x 2-center :( 0.032 s) ( 0.030 s)
| Transforming ovlp_3fn to ovlp_3KS : 0.008 s 0.008 s
| Total time for GW calculation : 40.156 s 40.109 s
| GW self-energy (analytic cont.) : 39.572 s 39.522 s
| Total time for solution of K.-S. equations : 0.060 s 0.229 s
| Total time for density update : 6.436 s 6.461 s
| Total time for mixing : 1.504 s 1.481 s
| Total time for Hartree multipole update : 0.260 s 0.254 s
| Total time for Hartree multipole sum : 1.604 s 1.572 s
| Total time for total energy evaluation : 0.040 s 0.007 s
Partial memory accounting:
| Residual value for overall tracked memory usage across tasks: 0.000000 MB (should be 0.000000 MB)
| Peak values for overall tracked memory usage:
| Minimum: 3.659 MB (on task 3 after allocating grid_partition)
| Maximum: 5.183 MB (on task 4 after allocating grid_partition)
| Average: 4.425 MB
| Largest tracked array allocation:
| Minimum: 2.303 MB (all_coords on task 3)
| Maximum: 3.319 MB (all_coords on task 4)
| Average: 2.814 MB
Note: These values currently only include a subset of arrays which are explicitly tracked.
The "true" memory usage will be greater.
Have a nice day.
------------------------------------------------------------
Spectral function
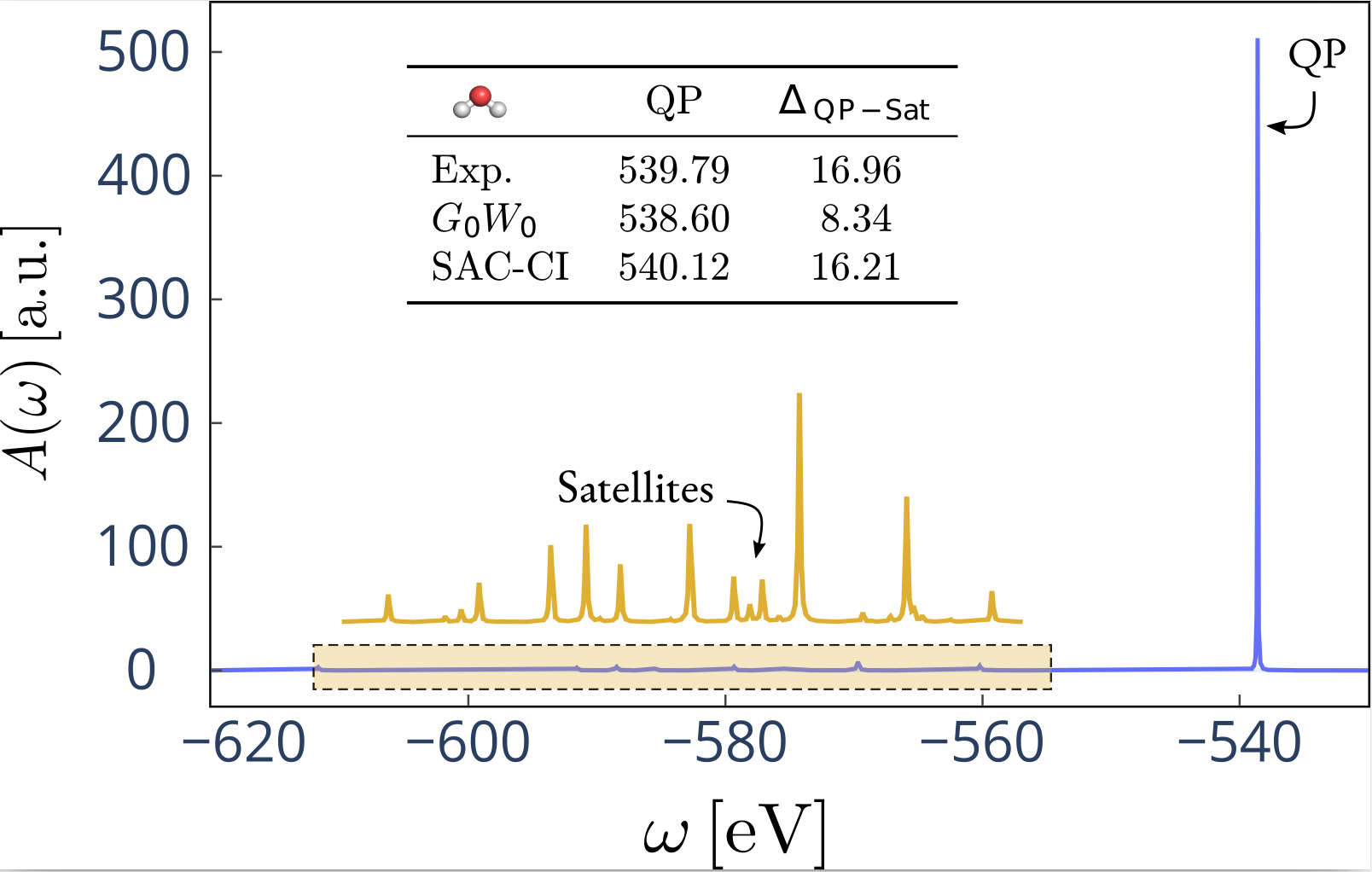
Learning resources
Publications and textbooks

|

|

|

|
Video tutorials
FHI-aims tutorial series
Thank you very much
Slides source on GitHub


Schwinger's functional derivative method
Define a generalized single-particle Green's function dependent of a probe field \(\varphi\), analyze its linear response with respect to the latter, and do a lot of algebra.
Taking the functional derivative of \(G(1,2)\) eventually leads to the Dyson equation, defining several quantities on the way: